Un importante insieme di reazioni chimiche, che differiscono dalla reazioni ioniche o radicaliche per molti aspetti, sono state riconosciute e studiate in modo approfondito. Tra le caratteristiche condivise da queste reazioni, tre in particolare le rendono diverse:
1. Esse sono relativamente inalterate dai cambi di solvente, dalla presenza di iniziatori radicalici o reagenti di scavenging, o (con qualche eccezione) da catalizzatori elettrofili o nucleofili.
2. Esse procedono attraverso una serie simultanea (concertata) di rotture e formazioni di legami in un singolo passaggio cinetico, spesso con elevata stereospecificità.
3. In accordo con 1 & 2, nessun intermedio ionico, radicalico o altro intermedio distinguibile si trovano lungo il cammino di reazione.
Dato che reazioni di questo tipo spesso procedono attraverso riorganizzazione pressoché simultanea di coppie di elettroni leganti tramite stati di transizione ciclici, esse sono state chiamate reazioni pericicliche.
Le quattro principali classi di reazioni pericicliche sono chiamate: Cicloaddizione, Elettrocicliche, Sigmatropiche, e reazioni Ene. Una illustrazione generale di ogni classe è mostrata negli schemi sottostanti. La cicloaddizione e le reazioni ene sono mostrate nel loro formato intermolecolare. Le corrispondenti reazioni intramolecolari, che creano un anello in più, sono ben note.
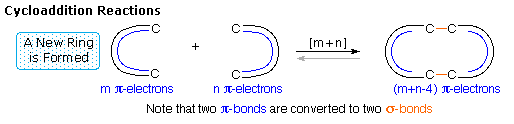
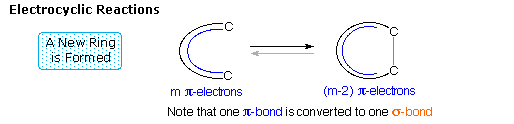
Tutte queste reazioni sono potenzialmente reversibili (notare le frecce grige). L'inverso di una cicloaddizione è chiamato cicloreversione e procede tramite rimozione di un abello e conversione di due legami sigma in due legami pi. La reazione elettrociclica mostrata sopra è un processo di formazione d'anello. L'inverso della reazione di apertura d'anello elettrociclica procede tramite conversione di un legame sigma in un legame pi. Come mostrato, la reazione retro ene spacca un composto insaturo in due frammenti insaturi. Infine, gli shift sigmatropici di legame possono coinvolgere semplici gruppi migranti, come mostrato nell'esempio sopra, o possono avvenire tra due sistemi di elettroni pi (riarrangiamento di Cope).
Le descrizioni generali mostrate sopra forniscono una base per la classificazione delle reazioni, ma cautela deve essere presa per assicurarsi che una data trasformazione sia davvero concertata. Sfortunatamente, questa non è una determinazione banale, spesso è richiesta una combinazione di marcatura isotopica e studi stereochimici per arrivare ad una conclusione plausibile. C'è anche una sottile distinzione che deve essere fatta tra una reazione sincrona in cui tutti gli eventi di formazione e rottura dei legami avvengono all'unisono, ed un processo concertato multi stadio in cui alcuni eventi precedono altri senza generare uno stato intermedio.
Sebbene alcune reazioni pericicliche avvengano spontaneamente, la maggior parte di essere richiedono l'introduzione di energia nella forma di radiazione UV/vis o calore, con un dipendenza notevole del prodotto dalla fonte di energia usata. Un apprezzamento dei cambiamenti strutturali stereospecifici che queste reazioni promuovono è ottenuto meglio tramite l'analisi di alcuni esempi individuali.
I seguenti utenti ringraziano quimico per questo messaggio: None
1. Reazioni di cicloaddizione
Una combinazione concertata di due sistemi π-elettronici per formare un anello di atomi avente due nuovi legami σ e meno di due legami π è chiamata reazione di cicloaddizione. Il numero degli elettroni π partecipanti per ogni componente è dato nella parentesi che precede il nome, e la riorganizzazione degli elettroni può essere illustrata da un ciclo di frecce curve - ognuna rappresentante il movimento di una coppia di elettroni.
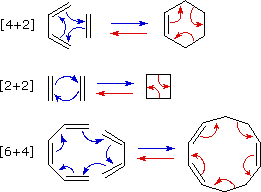
La reazione di cicloaddizione che forma l'anello è descritta da frecce blu, mentre il processo di cicloreversione che apre l'anello è designato con frecce rosse. Notate che il numero di frecce curve necessario a mostrare la riorganizzazione dei legami è la meta del numero totale tra parentese.
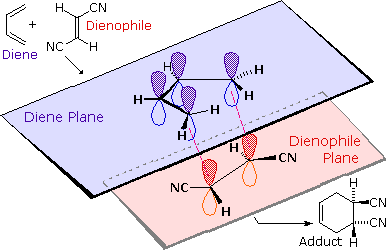
La più comune reazione di cicloaddizione è la ciclizzazione [4π+2π] nota come reazione di Diels-Alder. Nella terminologia della Diels-Alder ci si riferisce ai due reagenti usando i termini diene e dienofilo.
Lo schema sotto mostra due esempi di cicloaddizione [4π+2π], e nella seconda equazione una successiva cicloaddizione [2π+2π] indotta da luce. In ogni caso il diene è colorato in blu, ed i nuovi legami σ nell'addotto sono colorati in rosso. La stereospecificità di queste reazioni dovrebbe essere evidente.
Nel primo esempio, i sostituenti acetossi sul diene hanno identiche configurazioni E, ed essi rimangono cis l'uno rispetto all'altro nell'addotto ciclico. Ugualmente, i sostituenti esterei su dienofilo hanno una configurazione trans che è mantenuta nell'addotto.
I reagenti nella seconda equazione sono entrambi monociclici, così l'addotto di cicloaddizione ha tre anelli. L'orientazione dell'anello a sei membri chinone rispetto al sistema bicicloeptano (colorato in blu) è endo, che significa che esso è orientato cis rispetto al ponte più lungo o più insaturo. La configurazione alternativa è chiamata eso.
Dato che il dienofilo (chinone) ha due doppi legami attivati, una seconda reazione di cicloaddizione è possibile, purché sufficiente diene venga fornito. La seconda cicloaddizione è più lenta della prima, così il monoaddotto mostrato qui è facilmente preparato con buona resa. Sebbene quest prodotto [4+2] sia stabile al successivo riscaldamento, esso subisce una cicloaddizione [2+2] quando esposto alla luce del sole. Notate la perdita di due legami π carbonio-carbonio e la formazione di due legami σ (colorati in rosso) in questa trasformazione. Notate anche che il pedice π è spesso omesso dalla notazione [m+n] per la maggior parte della cicloaddizione che coinvolgono sistemi ad elettroni π.
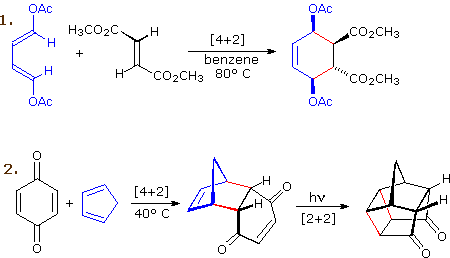
La reazione 3 è una reazione di Diels-Alder intramolecolare. Dato che diene e dienofilo sono uniti da una catena di atomi, la risultante cicloaddizione [4+2] di fatto forma due nuovi anelli, uno dalla cicloaddizione e l'altro dalla catena che collega. Ancora una volta l'addizione è stereospecifica, ignorando il sostituente isopropile, essendo la fusione cis ed endo. La quarta reazione è una cicloaddizione [6+4].
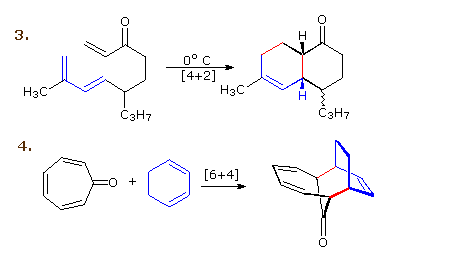
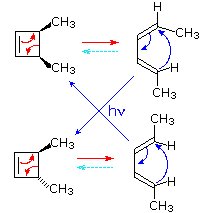
2. Reazioni elettrociclicheUna reazione elettrociclica è la ciclizzazione concertata di un sistema ad elettroni π coniugato tramite conversione di un legame π in un legame σ che forma l'anello. La reazione inversa può essere chiamata apertura d'anello elettrociclica.
Due esempi sono mostrati sotto. La chiusura d'anello elettrociclica è designata da frecce blu, e l'apertura d'anello da frecce rosse. Ancora una volta, il numero di frecce curve che descrivono la riorganizzazione dei legami è la metà del numer di elettroni coinvolti nel processo.
Nel primo caso, il trans,cis,trans-2,4,6-ottatriene subisce chiusura d'anello termica a cis-5,6-dimetil-1,3-cicloesadiene. La stereospecificità di questa reazione è dimostrata dalla chiusura dell'isomerico trans,cis,cis-triene a trans-5,6-dimetil-1,3-cicloesadiene, come notato nel secondo esempio.
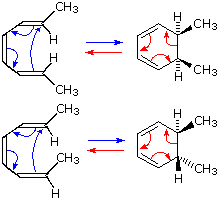
Nello schema sotto sono mostrati due esempi di apertura elettrociclica termica di ciclobuteni a butadieni coniugati. Questa modalità di reazione è favorita dall'allentamento della tensione d'anello, e l'inversa chiusura d'anello (frecce blu chiaro) non è di norma osservata. La chiusura d'anello fotochimica può essere effettuata, ma la stereospecificità è opposta rispetto all'apertura d'anello termica.
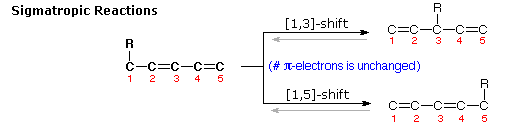
3. Riarrangiamenti sigmatropici
Riarrangiamenti molecolari in cui un atomo o un gruppo legato σ, affiancato da uno o più sistemi π elettronici, shifta verso una nuova posizione con una riorganizzazione corrispondente dei legami π sono chiamati riarrangiamenti sigmatropici.
Il numero totale di legami σ e di legami π rimane inalterato.
Questi riarrangiamenti sono descritti da due numeri messi tra parentesi, che si riferiscono alla distanza relativa (in atomi) verso cui ogni estremità del legame σ si è mossa, come illustrato dalla prima equazione nello schema sotto.
Il più comune atomo a subire shifts sigmatropici è l'idrogeno o uno dei suoi isotopi.
La seconda equazione nello schema mostra un semplice shift di idrogeno [1,5] che converte una relativamente stabile sistema allenico in un triene coniugato. Notate che questo riarrangiamento, che comporta la rilocazione di tre coppie di elettroni leganti, può essere descritto da tre frecce curve.
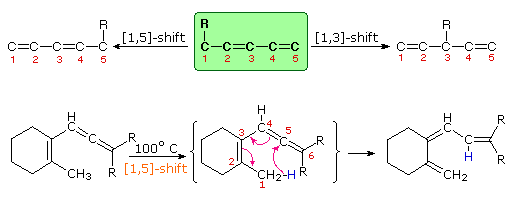 Nello schema sotto sono riportati altri due esempi di shift di idrogeno [1,5] termico. Queste reazioni sono particolarmente illustrative del fatto che shifts di idrogeno [1,3] non sono osservati.
Il reagente della prima equazione è un 1,3,5-cicloottatriene marcato con deuterio. Per riscaldamento, questo composto equilibria con il suo isomero 1,3,6-triene, ed i due atomi di deuterio sono scambiati tra le quattro posizione note. Se stessero avvenendo shifts di idrogeno [1,3] o [1,7], gli atomi di deuterio dovrebbero essere distribuiti equamente tra tutti gli otto atomi di carbonio. Per riscaldamento prolungato o a temperature maggiori questi cicloottatrieni subiscono apertura d'anello elettrociclica a 1,3,5,7-ottatetraene e richiusura a vinil-1,3-cicloesadieni.
Nello schema sotto sono riportati altri due esempi di shift di idrogeno [1,5] termico. Queste reazioni sono particolarmente illustrative del fatto che shifts di idrogeno [1,3] non sono osservati.
Il reagente della prima equazione è un 1,3,5-cicloottatriene marcato con deuterio. Per riscaldamento, questo composto equilibria con il suo isomero 1,3,6-triene, ed i due atomi di deuterio sono scambiati tra le quattro posizione note. Se stessero avvenendo shifts di idrogeno [1,3] o [1,7], gli atomi di deuterio dovrebbero essere distribuiti equamente tra tutti gli otto atomi di carbonio. Per riscaldamento prolungato o a temperature maggiori questi cicloottatrieni subiscono apertura d'anello elettrociclica a 1,3,5,7-ottatetraene e richiusura a vinil-1,3-cicloesadieni.
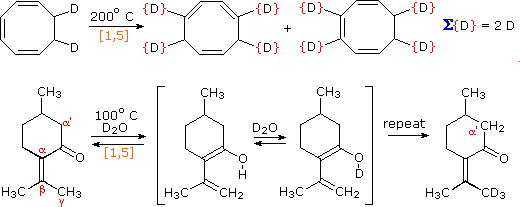 Il secondo esempio mostra un altro shift di idrogeno [1,5], dal gruppo metile prossimale all'atomo di ossigeno del carbonile. Il dienolo risultante rapidamente scambia OH con OD prima che lo shift [1,5] torni indietro. In questo modo il metile reattivo è presto convertito a CD3. Dato che gli idrogeni in alfa a gruppi carbonilici sono noti subire scambio acido o base catalizzato tramite intermedi enolici, dovremmo aspettarci che anche il gruppo α'-CH2 scambi. Comunque, se si fa attenzione nel rimuovere catalizzatori acidi o basici, si trova che lo shift termico [1,3] necessario per lo scambio è molto lento.
Il secondo esempio mostra un altro shift di idrogeno [1,5], dal gruppo metile prossimale all'atomo di ossigeno del carbonile. Il dienolo risultante rapidamente scambia OH con OD prima che lo shift [1,5] torni indietro. In questo modo il metile reattivo è presto convertito a CD3. Dato che gli idrogeni in alfa a gruppi carbonilici sono noti subire scambio acido o base catalizzato tramite intermedi enolici, dovremmo aspettarci che anche il gruppo α'-CH2 scambi. Comunque, se si fa attenzione nel rimuovere catalizzatori acidi o basici, si trova che lo shift termico [1,3] necessario per lo scambio è molto lento.
I riarrangiamenti [3,3] sigmatropici di 1,5-dieni o allil vinil eteri, noti rispettivamente come
riarrangiamenti di Cope e Claisen, sono tra le reazioni sigmatropiche più comunemente usate.
Tre esempi del riarrangiamento di Cope sono mostrati nel seguente schema.
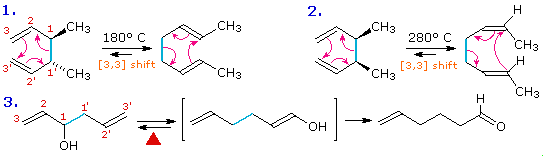
Le reazioni 1 e 2 (fila in alto) dimostrano la stereospecificità di questa reazione. I legami σ blu chiaro legano i due gruppi allile, orientati così che le loro estremità siano vicine l'una all'altra. Dato che ogni segmenti allilico è il luogo dello shift [1,3], la reazione totale è classificata come riarrangiamento [3,3]. Le tre frecce curve colorate in rosa descrivono la ridistribuzione delle tre coppie elettroniche leganti nel corso di questo riarrangiamento reversibile.
Il diene che reagisce nella terza reazione è disegnato in una conformazione estesa. Questa molecola deve assumere una conformazione a spirale (come sopra) prima che il riarrangiamento [3,3] possa avvenire. Il prodotto di questo riarrangiamento è un enolo che immediatamente tautomerizza alla sua forma chetonica. Tali varianti sono denominate come
riarrangiamento ossi-Cope, e sono utili perché il riarrangiamento inverso è bloccato dalla rapida chetonizzazione. Se il sostituente idrossile è convertito ad un sale di alcossido, l'energia di attivazione del riarrangiamento è diminuita in modo significativo.
Il riarrangiamento di Cope degenere o auto-replicante è stata un soggetto affascinante di ricerca. Ma di questo ne parlerò magari più avanti, sempre qui, per la gioia di rock.angel

.
La reazione 4 è un classico riarrangiamento di un allil fenil etere ad
o-allil fenolo. Il sostituente metile sulla funzionalità allile serve a dimostrare lo shift di legame a questo sito. L'iniziale prodotto cicloesadienone immediatamente tautomerizza a fenolo, riottenendo la stabilità dell'anello aromatico.
La reazione 5 è un analogo alifatico in cui un gruppo vinile sostituisce l'anello aromatico.
In entrambi i casi tre coppie di elettroni leganti subiscono una riorganizzazione.
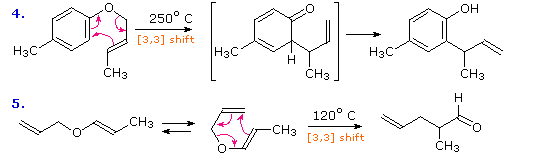
Nello schema sotto sono riportati due esempi di riarrangiamenti [2,3] sigmatropici.
Il solfossido allilico nella reazione 6 riarrangia reversibilmente ad un meno stabile estere sulfenato. Il debole legame S-O può essere rimosso riduttivamente da trimetil fosfito ad un alcole allilico e ad un tiolo (non mostrato).
La reazione 7 mostra un riarrangiamento simile di una ilide di zolfo a solfuro ciclico. Il riarrangiamento [2,3] di Wittig è ancora un altro esempio.
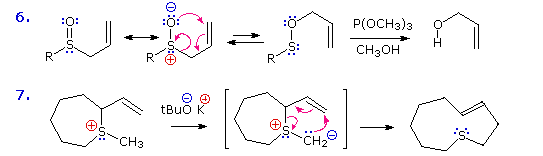
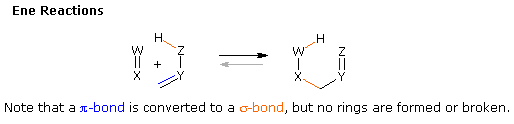
4. Reazioni ene
L'unione di un doppio o un triplo legame ad un alchene avente un idrogeno allilico trasferibile è chiamata reazione ene. Il processo inverso è chiamato reazione retro ene. Nelle direzione che porta al legame la reazione ene è caratterizzata dalla ridistribuzione di tre coppie di elettroni leganti e può essere descritta da un ciclo di tre frecce curve. Come notato prima, questa riorganizzazione del legame comporta la conversione totale di un legame π in un legame σ (o l'opposto nel caso di una frammentazione retro ene). Questo è lo stesso cambiamento di legame contabile esibito dalle reazioni elettrocicliche, non vengono formati o rotti anelli in una reazione ene a meno che non sia intramolecolare.
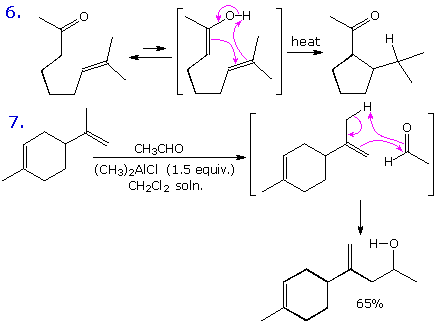
I seguenti esempi illustrano alcune tipiche reazioni ene; l'equazione 3 è una reazione ene intramolecolare.
Le reazioni ene sono favorite quando il reagente che accetta l'idrogeno, o "enofilo", è elettrofilo. Questo è il caso per le reazioni 1 e 2, che avvengono in condizioni più blande della reazione 3, nonostante la natura intramolecolare dell'ultima.
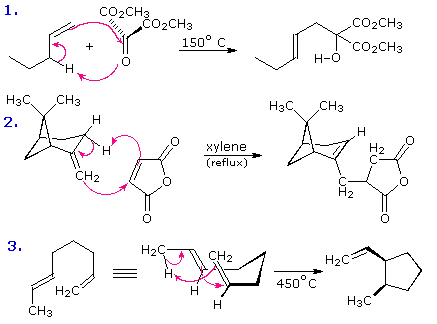
L'idrogeno è il più comune atomo trasferito in una reazione ene. Inoltre, tutti gli esempi mostrati sopra comportano shifts di idrogeno.
Altri atomi o gruppi possono, comunque, partecipare in trasformazioni tipo ene. Due casi di questo genere sono mostrati sotto.
La reazione 4 è disegnata come una reazione retro ene, sebbene questa non abbia dimostrato di essere un esempio generale per tutte le reazioni di alcoli allilici con tionile cloruro.
L'equazione 5 illustra un'insolita "reazione ene con magnesio" in cui una funzione Grignard si muove verso una nuova posizione prima di reagire con un reagente elettrofilo come la CO2. Poiché questa è una reazione ene intramolecolare, un nuovo anello viene formato.
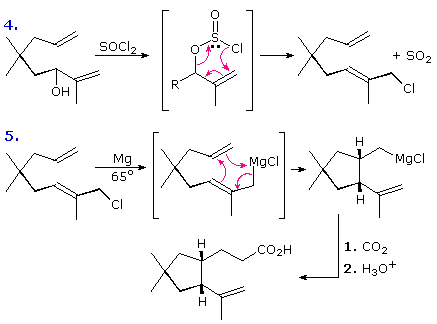
Sotto sono mostrati altri due esempi.
L'equazione 6 dimostra che un enolo tautomero, anche in bassa concentrazione, può funzionare come donatore di idrogeno nella reazione ene.
L'equazione 7 è uno dei molti esempi di catalisi di acido di Lewis nella reazione ene. Una simile reazione acido catalizzata di semplici aldeidi con alcheni a dare alcoli allilici, 1,3-dioli o 1,3-diossani è nota come reazione di Prins.
Alcune reazioni retro ene si sono dimostrate utili come trasformazioni sintetiche. Ma non ne parlerò ora... ma sarà oggetto magari di un altro topic 
5. Notazioni stereochimiche
Una caratteristica condivisa dalla maggior parte delle reazioni pericicliche, e notata in molti casi descritti sopra, è la loro stereospecificità.
Questa non è la prima classe di reazioni per cui è stata notata una caratteristica stereospecificità.
Le reazioni di sostituzione possono procedere casualmente o tramite "inversione" o "ritenzione" di configurazione.
Le reazioni di eliminazione possono avvenire in un modo "anti" o "syn", o essere configurazionalmente casuali. Il termini "syn" e "anti" sono stati anche applicati a reazioni di addizione 1,2.
Dato che queste notazioni di cambio configurazionale non sono appropriate per le reazioni pericicliche, sono necessarie nuove designazioni.
Le reazioni di cicloaddizione e i riarrangiamenti sigmatropici coinvolgono entrambi coppie di eventi di formazione di legami σ (o una accoppiata di formazione & rottura di legame) associate con un sistema ad elettroni π. Se tutti gli eventi di legame avvengono sulla stessa faccia del sistema π la configurazione della reazione è denominata suprafacciale. Se gli eventi di legame avvengono sui lati opposti o sulle facce del sistema π la reazioni è denominata antarafacciale.
Esempi suprafacciali di queste trasformazioni pericicliche sono mostrati sotto. I numeri tra parentesi che designano le reazioni di questo tipo spesso contengono pedici (s o a) che specificano questa configurazione. Perciò la cicloaddizione a sinistra può essere denominato un processo [4s + 2s].
Sebbene le reazioni di cicloaddizione siano concertate (nessuna specie intermedia si forma), i due nuovi legami non sono vengono necessariamente formati in una maniera sincrona. A seconda della parziale distribuzione di carica nel diene e nel dienofilo, la formazione di un legame può portare allo sviluppo dell'altro. Tali stati di transizione di legame asimmetrici sono denominati asincroni.
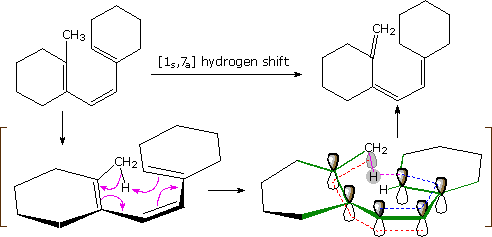
Un esempio di uno shift di idrogeno [1,7] antarafacciale è mostrato nello schema seguente. Il triene coniugato assume una conformazione quasi planare a spirale in cui un idrogeno del metile è orientato giusto sopra la fine dell'atomo di carbonio dell'ultimo doppio legame. Uno shift di idrogeno [1s,7a] sigmatropico può quindi avvenire, come descritto dalle quattro frecce curve. In riferimento al piano approssimato di questo sistema π elettronico (definito da legami verdi), l'atomo di idrogeno si allonta dalla faccia inferiore e lega sulla faccia superiore, così il trasferimento è antarafacciale.
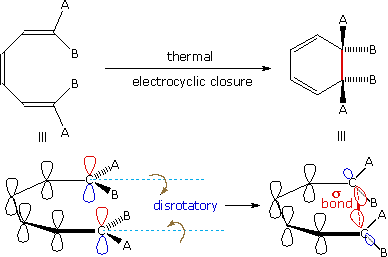
Una notazione differente per il cambiamento configurazionale è richiesta per le reazioni elettrocicliche. In questi casi il legame σ tra le estremità del sistema π coniugato è o creato o rotto con una corrispondente perdita o guadagno di un legame π. Affinché questo avvenga, gli atomi di carbonio terminali del sistema π coniugato devono essere reibridizzati con una rotazione o avvitamento di accompagnamento di approssimativamente 90º. Quando visti lungo l'asse di rotazione, i due gruppi terminali possono girare nella stessa direazione (conrotatori), o in direzioni opposte (disrotatori). I prefissi con e dis possono essere ricordati per associazione con la loro presenza in parole come concordare & dissentire. Questi due modi di reazione elettrociclica sono mostrati nel diagramma sotto nella forma generale in cui essi sono più comunemente osservati.
Dato che le reazioni ene di solito comportano operazioni accoppiate di formazione di legame & di rottura di legame associate a corti sistemi π (2 o 3 carboni), la loro stereospecificità è quasi sempre suprafacciale rispetto ad entrambi i componenti. Questa caratteristica configurazionale è illustrata dalla equazione retro ene sotto.
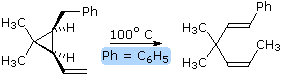
Qui sotto, è mostrata una rappresentazione dello stato di transizione per questa trasformazione stereospecifica. Notate che la rottura e la formazione del legame avvengono in una orientazione suprafacciale rispetto ad ogni sistema π. Questa reazione è facilitata dall'allentamento della tensione del piccolo anello.
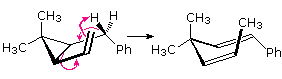
Diverse differenti relazioni strutturali tra le metà reagenti di una reazione ene intramolecolare sono possibili. Gli esempi mostrati qui e sopra rappresentano la maggior parte della orientazione comune.
6. Caratteristiche sconcertanti delle reazioni pericicliche
Gli esempi delle reazioni pericicliche presentati qui forniscono un'ampia prova della loro utilità nel costruire o modificare molecole complesse, spesso con un elevato grado di stereospecificità. Comunque, in contrasto con l'applicabilità generale della maggior parte delle comuni reazioni ioniche, le reazioni pericicliche spesso mostrano una marcata sensibilità ai piccoli cambiamenti strutturali. Perciò, la stereospecificità può fare marcia indietro, e le velocità possono variare un milione di volte o più.
Nel caso delle reazioni di cicloaddizione, le tre equazioni sotto riportate illustrano questo fatto.
Le equazioni 1 e 2 mostrano due trasformazioni molto simili, ma la prima avviene con moderato riscaldamento e la seconda no. Notate che in ogni caso il triplo legame contribuisce con solo elettroni allo stato di transizione della cicloaddizione. La comune cicloaddizione [4+2] nota come reazione di Diels-Alder procede stereospecificamente in maniera suprafacciale, ma la cicloaddizione [14+2] nell'equazione 3 è antarafacciale rispetto al poliene.
Le reazioni elettrocicliche e sigmatropiche mostrano anche enigmatiche differenze in comportamento.
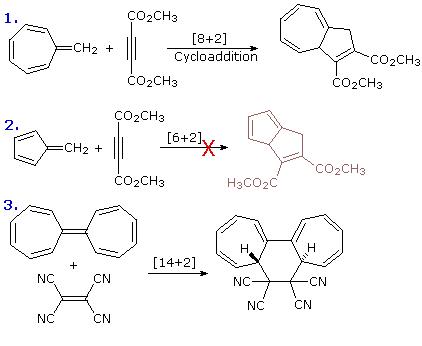
Nello schema sotto sono mostrati altri quattro esempi.
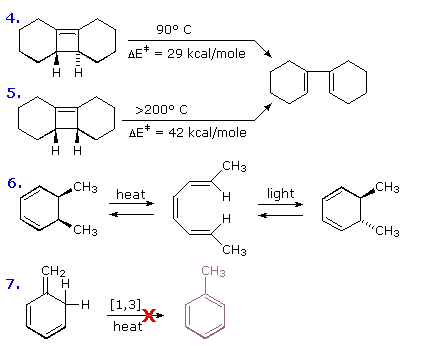
Le equazioni 4 e 5 descrivono aperture di anello elettrocicliche simili di ciclobuteni stereoisomerici. La prima avviene in condizioni di riscaldamento relativamente blande, ma la seconda richiede calore estremo e può procedere bene tramite omolisi del legame ad un diradicale.
L'equazione 6 mostra due chiusure d'anello elettrocicliche del trans,cis,trans-2,4,6-ottatriene. La reazione termica è disrotatoria, ed il processo fotochimico è conrotatorio.
Infine, l'assenza di shifts [1,3] sigmatropici di idrogeno era stata notata in precedenza, ed un chiaro esempio è mostrato nell'equazione 7. L'isomerizzazione del triene coniugato a toluene dovrebbe essere fortemente esotermica, ma un riarrangiamento concertato di questo tipo dovrebbe essere un processo [1,3] sigmatropico. In assenza di catalizzatori acidi questo triene è completamente stabile al riscaldamento moderato. Qualsiasi shift [1,5] di idrogeno che avviene ricostruisce il triene di partenza e richiederebbe una marcatura isotopica per provarlo. Naturalmente, l'isomerizzazione a toluene avviene rapidamente se dell'acido viene aggiunto.
7. Un'utile regola mnemonica
Prima che le reazioni pericicliche possano essere messe per l'uso in una maniera predicibile e controllata, deve essere formulata un'ampia comprensione meccanicistica dei fattori che influenzano queste trasformazioni concertate.
Il più semplice, sebbene il meno rigoroso, metodo per predirre il cammino configurazionale favorito da una proposta reazione periciclica è basato sul conto degli elettroni dello stato di transizione. Nella maggior parte degli esempi precedenti, le reazioni pericicliche erano state descritte da un ciclo di frecce curve, ognuna rappresentante una coppia di elettroni leganti. In numero totale degli elettroni che subiscono riorganizzazione è sempre pari, ed è o 4n+2 o 4n (dove n è un intero). Una volta che questo conto degli elettroni è fatto, la seguente tabella può essere usata per le predizioni.
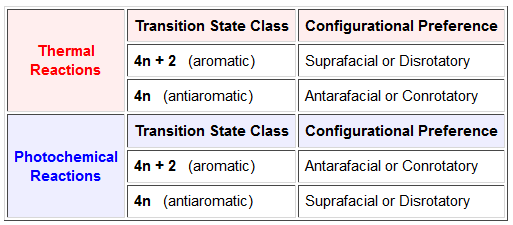
Sebbene questo modesto mezzo mnemonico non renda esplicito l'uso di orbitali molecolari, metodi più rigorosi che sono fondati sulle caratteristiche di tali orbitali hanno fornito un'importante visione in queste reazioni. Dato che le reazioni pericicliche procedono tramite riorganizzazione ciclica di coppie di elettroni leganti, è necessario valutare i cambiamenti negli orbitali molecolari associati che avvengono nel cammino dai reagenti ai prodotti.
Ma per il momento non parlerò di queste cose. Anche perché è un discorso lungo e complesso. Qualora servisse sono disposto a metterlo.
Riarrangiameti di Cope auto-replicanti
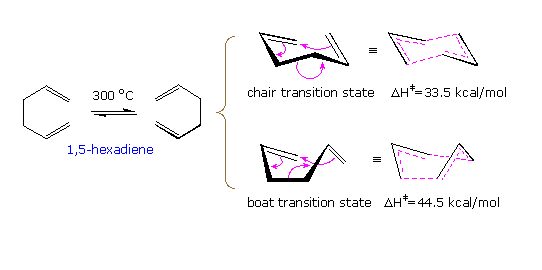
Il più semplice riarrangiamento di Cope è il riarrangiamento degenere dell'1,5-esatriene. Come disegnato nello schema seguente, questo shift [3,3]-sigmatropico può avvenire tramite uno dei due stati di transizione, uno stato a sedia a minore energia o uno stato a barca a maggiore energia. Dato che il prodotto è identico al reagente, è necessaria la marcatura isotopica per stabilire quale cambiamento è avvenuto. L'energia di attivazione relativamente alta di questa reazione richiede una temperatura vicina ai 300 ºC.
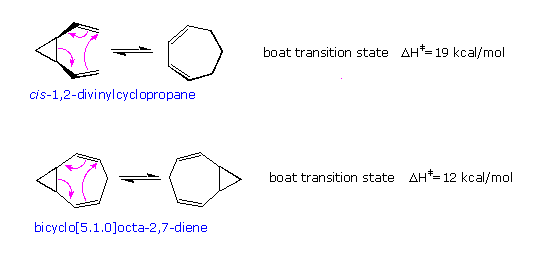
 Se la tensione d'anello è alleviata in questo riarrangiamento, come nel caso del cis-1,2-divinilciclopropano, l'energia di attivazione è sostanzialmente ridotta così che la conversione a 1,4-cicloeptadiene avviene sotto i 20 ºC in 30 minuti, come mostrato. Questo riarrangiamento diventa degenere per il composto biciclo[5.1.0]otta-2,7-diene, e l'energia di attivazione è ulteriormente ridotta. In entrambi i casi uno stato di transizione a barca è forzato dalla tensione strutturale.
Se la tensione d'anello è alleviata in questo riarrangiamento, come nel caso del cis-1,2-divinilciclopropano, l'energia di attivazione è sostanzialmente ridotta così che la conversione a 1,4-cicloeptadiene avviene sotto i 20 ºC in 30 minuti, come mostrato. Questo riarrangiamento diventa degenere per il composto biciclo[5.1.0]otta-2,7-diene, e l'energia di attivazione è ulteriormente ridotta. In entrambi i casi uno stato di transizione a barca è forzato dalla tensione strutturale.
 Se si formano analoghi triciclici tramite ponte C1-C8 del biciclo[5.1.0]otta-2,7-diene, si osservano simili riarrangiamenti degeneri.
Se si formano analoghi triciclici tramite ponte C1-C8 del biciclo[5.1.0]otta-2,7-diene, si osservano simili riarrangiamenti degeneri.
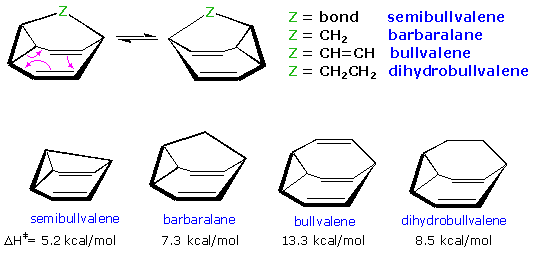 Per tre di questi composti l'energia di attivazione è ancora minore a causa della combinazione dell'aumentata tensione allo stato fondamentale e della stabilizzazione dello stato di transizione tramite legame interallilico.
L'idrocarburo C10H10, o bullvalene, è degno di nota ed eccezionale per due aspetti. Primo, il suo stato fondamentale è stabilizzato rispetto all'analogo diidro tramite coniugazione π con l'anello a tre membri. Secondo, la sua simmetrica tripla permette numerosi riarrangiamenti di Cope auto-replicanti che portano ad più di un milione (10!/3=1,209,600) di tautomeri con identica valenza. Tutti i carboni e gli idrogeno nel bullvalene sembrano essere equivalenti sulla scala dei tempi all'NMR a 100 ºC, e non possono essere discriminati sino a quando non si raffredda a -15 ºC. Molecule di questo tipo sono denominate flussionali. È importante riconoscere che la tautomerizzazione della valenza di questo tipo non è risonanza (le posizione relative degli atomi subiscono piccolo cambiamenti).
Per tre di questi composti l'energia di attivazione è ancora minore a causa della combinazione dell'aumentata tensione allo stato fondamentale e della stabilizzazione dello stato di transizione tramite legame interallilico.
L'idrocarburo C10H10, o bullvalene, è degno di nota ed eccezionale per due aspetti. Primo, il suo stato fondamentale è stabilizzato rispetto all'analogo diidro tramite coniugazione π con l'anello a tre membri. Secondo, la sua simmetrica tripla permette numerosi riarrangiamenti di Cope auto-replicanti che portano ad più di un milione (10!/3=1,209,600) di tautomeri con identica valenza. Tutti i carboni e gli idrogeno nel bullvalene sembrano essere equivalenti sulla scala dei tempi all'NMR a 100 ºC, e non possono essere discriminati sino a quando non si raffredda a -15 ºC. Molecule di questo tipo sono denominate flussionali. È importante riconoscere che la tautomerizzazione della valenza di questo tipo non è risonanza (le posizione relative degli atomi subiscono piccolo cambiamenti).
Classi di reazioni ene intramolecolari
Le metà "ene" (C=C–Z–H) ed "enofilica" (X=Y) di una reazione ene intramolecolare possono assumere differenti orientazioni relative che dipendono dalla natura della struttura legante.
La più comuna relazione è il
Tipo I, in cui l'enofilo è unito al carbonio dell'alchene più lontano dal gruppo Z–H. Le reazioni del
Tipo II hanno l'enofilo unio al carbonio dell'alchene che porta il gruppo Z–H, nelle reazioni del
Tipo III l'enofilo è unito direttamente all'atomo Z.
Questi diversi riarrangiamenti sono definiti nello schema seguente, dove gli atomi X,Y & Z sono colorati in blu, e l'idrogeno trasferito è in verde.
La maggior parte delle reazioni ene, inclusi gli esempi precedenti, sono di
Tipo I.
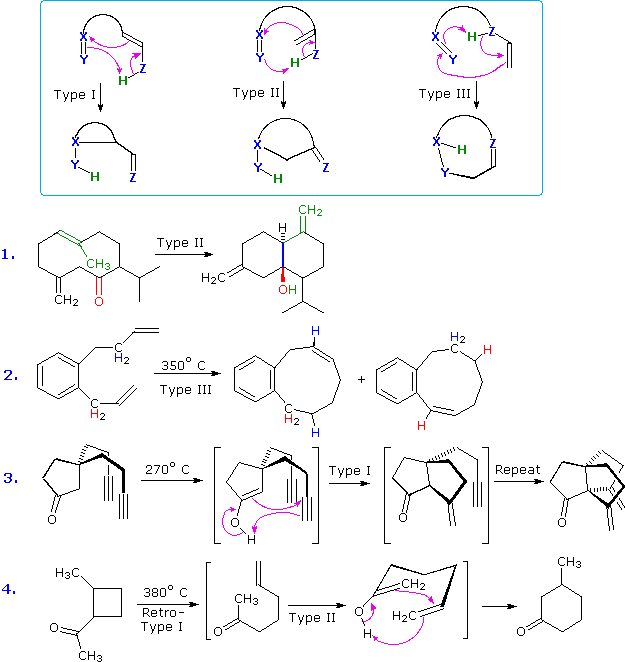
L'equazione 1 dimostra una reazione ene di Tipo II in cui l'enofilo è un gruppo carbonilico (colorato in rosso). La metà alchenica è colorata in verde, ed il nuovo legame sigma in blu.
L'equazione 2 è un raro esempio di una reazione ene di Tipo III. Ci sono di fatto due diverse reazioni ene che avvengono, e queste possono essere distinte tramite la locazione del doppio legame del prodotto e l'origine dell'atomo di idrogeno trasferito (colorati in rosso e blu nell'illustrazione sopra).
Gli ultimi due esempi mostrati mostrano un'interessante variante della reazione ene in cui il tautomero enolo di una funzione carbonile serve da componente "ene", ed un doppio o triplo legame carbonio-carbonio è l'"enofilo".
La reazione 3 ha due catene alchiniche orientante in modo adeguato per una reazione di Tipo I, ed entrambe partecipano sequenzialmente portando al nuovo composto triciclico "propellano".
Ci sono due trasformazione ene nell'equazione 4. La prima è una reazione retro ene di Tipo I, facilitata dall'alleviamento della tensione d'anello. La seconda è una ciclizzazione ene di Tipo II.
Una chiusura d'anello alternativa di Tipo III ad 1-metil-3-cicloesenolo non avviene.