quimico
2012-01-04 10:03
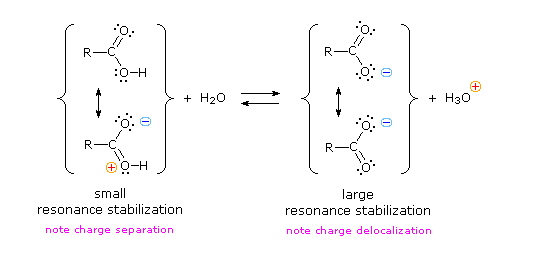
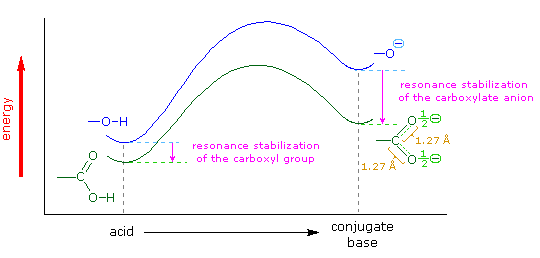
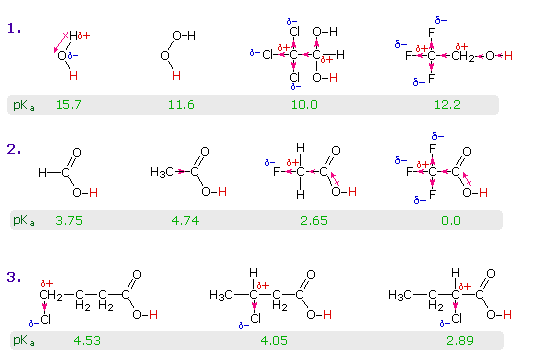
Il gruppo funzionale carbossile che caratterizza gli acidi carbossilici è insolito in quando esso è composti da due gruppi funzionali descritti in precedenza. Come potete vedere nella formula sotto, il gruppo carbossilico è costituito da un gruppo idrossile legat ad un gruppo carbonilico.
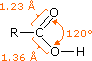
Esso è spesso scritto in forma condensata come –CO2H o –COOH. Altre combinazione di gruppi funzionali sono state descritte in precedenza, e sono stati descritti dei cambiamenti significativi nel comportamento chimico come risultato di interazioni di gruppo (ad es. fenolo & anilina). In questo caso, il cambiamento nelle proprietà chimiche e fisiche che risulta dall'interazione del gruppo idrossile e carbonile è così profondo che la combinazione è trattata di norma com un gruppo funzionale diverso e distinto.
1. Nomenclatura
Come con le aldeidi, il gruppo carbossile deve essere posizionato alla fine di un catena di carboni. Nel sistema di nomenclatura IUPAC il carbonio carbossilico è designato come #1, e gli altri sostituenti sono posizionati e nominati di conseguenza. Il suffisso IUPAC caratteristico per un gruppo carbossile è "acido -oico", e deve essere fatta attenzione nel non confondere questa nomenclatura sistematica con il simile sistema di nomenclatura comune. Queste due nomenclature sono illustrate nella seguente tabela, assieme ai loro m.p. e b.p.:
HCO2H
Nome comune: acido formico
Fonte: formiche (dal latino formica)
Nome IUPAC: acido metanoico
m.p.: 8.4 ºC b.p.: 101 ºC
CH3CO2H
Nome comune: acido acetico
Fonte: aceto (dal latino acetum)
Nome IUPAC: ethanoic acid
m.p.: 16.6 ºC b.p.: 118 ºC
CH3CH2CO2H
Nome comune: acido propionico
Fonte: latte (dal greco proteus prion)
Nome IUPAC: acido propanoico
m.p.: -20.8 ºC b.p.: 141 ºC
CH3(CH2)2CO2H
Nome comune: acido butirrico
Fonte: burro (dal latino butyrum)
Nome IUPAC: acido butanoico
m.p.: -5.5 ºC b.p.: 164 ºC
CH3(CH2)3CO2H
Nome comune: acido valerico
Fonte: radice di valeriana
Nome IUPAC: acido pentanoico
m.p.: -34.5 ºC b.p.: 186 ºC
CH3(CH2)4CO2H
Nome comune: acido caproico
Fonte: capre (dal latino caper)
Nome IUPAC: acido esanoico
m.p.: -4.0 ºC b.p.: 205 ºC
CH3(CH2)5CO2H
Nome comune: acido enantico
Fonte: vite (dal greco oenanthe)
Nome IUPAC: acido eptanoico
m.p.: -7.5 ºC b.p.: 223 ºC
CH3(CH2)6CO2H
Nome comune: acido caprilico
Fonte: capre
Nome IUPAC: acido ottanoico
m.p.: 16.3 ºC b.p.: 239 ºC
CH3(CH2)7CO2H
Nome comune: acido pelargonico
Fonte: pelargonio (un'erba)
Nome IUPAC: acido nonanoico
m.p.: 12.0 ºC b.p.: 253 ºC
CH3(CH2)8CO2H
Nome comune: acido caprico
Fonte: capre
Nome IUPAC: acido decanoico
m.p.: 31.0 ºC b.p.: 219 ºC
Gli acidi carbossilici sostituiti sono chiamati o tramite il sistema IUPAC o tramite i nomi comuni. Alcuni nomi comuni, per esempio l'aminoacido treonina, non hanno alcuna origine sistematica e devono essere semplicemente memorizzati. In altri casi, i nomi comuni fanno uso della notazione con lettere greche per gli atomi di carbonio vicini al gruppo carbossile. Alcuni esempi di entrambe le nomenclature sono forniti sotto.
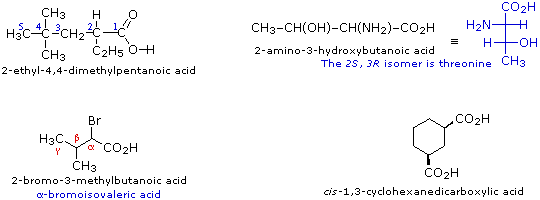
Gli acidi dicarbossilici semplici aventi la formula generale HO2C–(CH2)n–CO2H (dove n = 0-5) sono noti tramite nomi comuni: Acidi Ossalico (n = 0), Malonico (n = 1), Succinico (n = 2), Glutarico (n = 3), Adipico (n = 4) e Pimelico (n = 5). I nomi comuni, come questi, possono essere problematici da ricordare, così spesso si ricorre ad aiuti o trucchi mnemonici.
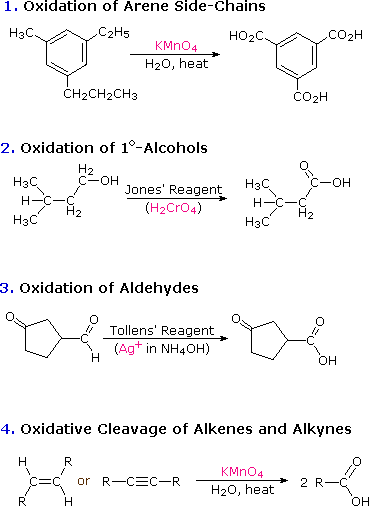
2. Prodotti naturali contenenti la funzione acido carbossilico
Gli acidi carbossilici sono diffusi in natura, spesso combinati con altri gruppi funzionali. Gli acidi carbossilici alchilici semplici, composti da quattro fino a dieci atomi di carbonio, sono liquidi o solidi bassofondenti aventi odori davvero sgradevoli. Gli acidi grassi sono componenti importanti delle biomolecole note come lipidi, specialmente grassi ed oli. Questi acidi carbossilici a catena lunga sono di solito chiamati con i loro nomi comuni, che nella maggior parte dei casi riflettono le loro fonti.
Cosa interessante, le mole cole della maggior parte degli acidi grassi naturali hanno un numero pari di atomi di carbonio. I composti analoghi con numeri dispari di atomi di carbonio sono perfettamente stabili e sono stati sintetizzati. Dato che la natura crea questi acidi a lunga catena unendo assieme unità acetato, non sorprende il fatto che gli atomi di carbonio che compongono i prodotti naturali siano multipli di due. I doppi legami nei composti saturi sono tutti cis (o Z).
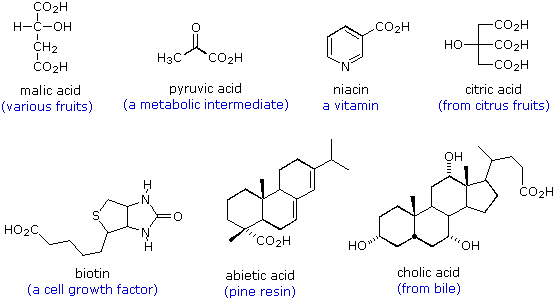
Le formule seguenti sono esempi di altri acidi carbossilici che si ritrovano in natura. Le strutture molecolari variano da semplici a complesse, spesso incorporano una varietà di altri gruppi funzionali, e molti sono chirali.
3. Derivati carbonilici correlati
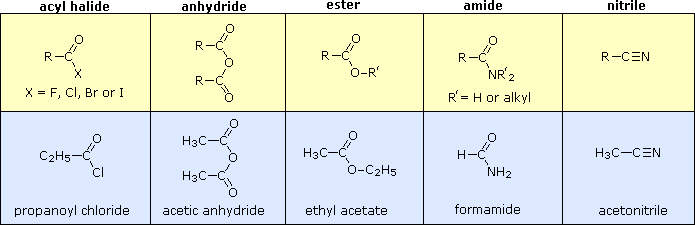
Altre combinazioni di gruppi funzionali con il gruppo carbonile possono essere preparate dagli acidi carbossilici, e solitamente sono trattati come derivati correlati ad essi. Nella tabella seguente sono elencate cinque classi comuni di questi derivati degli acidi carbossilici. Sebbene i nitrili non abbiano un gruppo carbonile, essi vengono inclusi qui perché gli atomi di carbonio della funzione hanno lo stesso stato di ossidazione. La fila superiore (giallo ombreggiato) mostra la formula generale per ogni classe, e la fila inferiore (blu chiaro) riporta un nome specifico per ognuna classe. Come nel caso delle amine, le ammidi sono classificate con primarie, secondarie e terziari, a seconda del numero di gruppi alchile legati all'azoto.
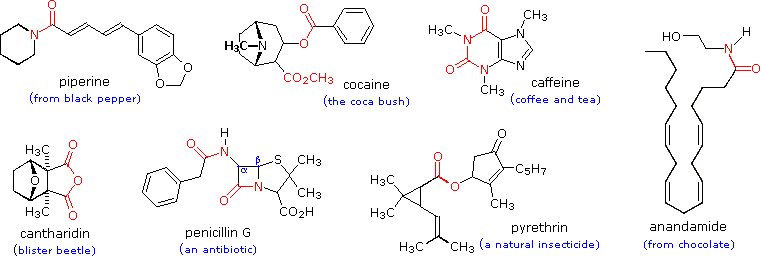
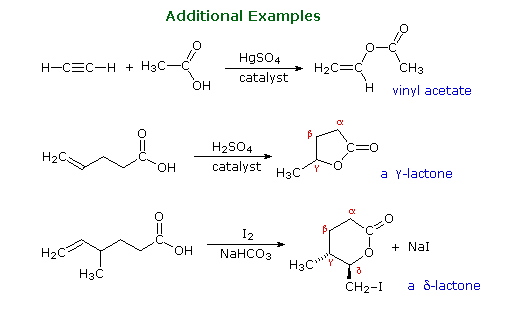
Gruppi funzionali di questo genere sono ritrovati in molti generi di prodotti naturali. Alcuni esempi sono mostrati sotto con il gruppo funzionale colorato in rosso. La maggior parte delle funzioni sono ammidi o esteri, e la cantaridina è un raro esempi di una anidride naturale. Gli esteri ciclici sono chiamati lattoni, e le ammidi cicliche sono chiamate lattami. La penicillina G ha due funzioni ammidiche, una delle quali è un β-lattame. La lettera greca posiziona l'azoto rispetto al gruppo carbonilico della ammide.
I seguenti utenti ringraziano quimico per questo messaggio: NaClO
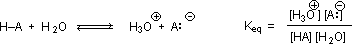
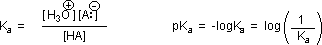
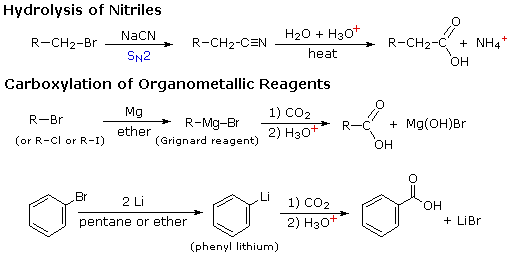
 RCO2–Na+ + CO2 + H2O
RCO2–Na+ + CO2 + H2O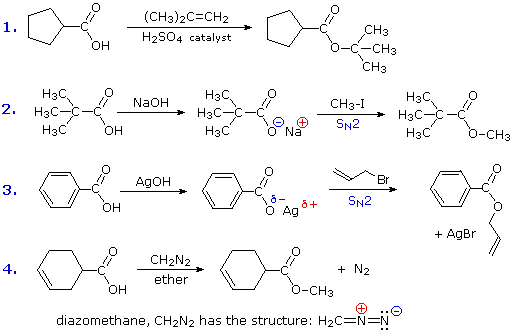
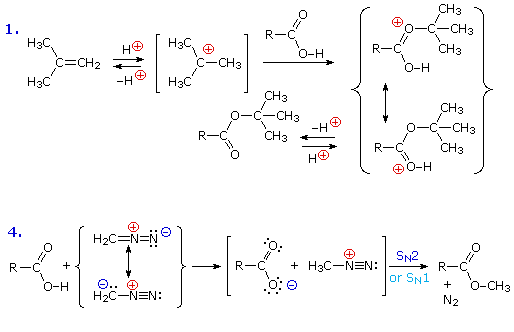
 La reazione #4 è chiamata esterificazione, poiché essa è comunemente usata per convertire gli acidi carbossilici nei loro derivati esteri. Gli esteri possono essere preparati in molti modi diversi; inoltre, le equazioni #1 e #4 nello schema precedente illustrano la formazione di t-butil e metil esteri rispettivamente. La formazione acido-catalizzata dell'etile acetato dall'acido acetico e dall'etanolo mostrata qui è reversibile, con una costante di equilibrio vicino a 2. La reazione può essere forzata a completezza rimuovendo l'acqua non appena è formata. Questo tipo di esterificazione è spessa chiamata esterificazione di Fischer. Come previsto, la reazione inversa, l'idrolisi acido-catalizzata dell'estere, può essere portata a termine aggiungendo un eccesso di acqua.
Un esame ponderato di questa reazione (#4) ci porta ad una domanda: perché essa è classificata come una sostituzione dell'idrossile piuttosto che una sostituzione dell'idrogeno? Le equazioni seguenti, in cui l'atomo di ossigeno dell'idrossile dell'acido carbossilico è colorato in rosso e quello dell'alcole in blu, illustrano questa distinzione (notate che i composti di partenza sono nel centro).
H2O + CH3CO-OCH2CH3 (H-sostituzione)
La reazione #4 è chiamata esterificazione, poiché essa è comunemente usata per convertire gli acidi carbossilici nei loro derivati esteri. Gli esteri possono essere preparati in molti modi diversi; inoltre, le equazioni #1 e #4 nello schema precedente illustrano la formazione di t-butil e metil esteri rispettivamente. La formazione acido-catalizzata dell'etile acetato dall'acido acetico e dall'etanolo mostrata qui è reversibile, con una costante di equilibrio vicino a 2. La reazione può essere forzata a completezza rimuovendo l'acqua non appena è formata. Questo tipo di esterificazione è spessa chiamata esterificazione di Fischer. Come previsto, la reazione inversa, l'idrolisi acido-catalizzata dell'estere, può essere portata a termine aggiungendo un eccesso di acqua.
Un esame ponderato di questa reazione (#4) ci porta ad una domanda: perché essa è classificata come una sostituzione dell'idrossile piuttosto che una sostituzione dell'idrogeno? Le equazioni seguenti, in cui l'atomo di ossigeno dell'idrossile dell'acido carbossilico è colorato in rosso e quello dell'alcole in blu, illustrano questa distinzione (notate che i composti di partenza sono nel centro).
H2O + CH3CO-OCH2CH3 (H-sostituzione)  CH3CO-OH + CH3CH2-OH
CH3CO-OH + CH3CH2-OH  (HO-sostituzione) CH3CO-OCH2CH3 + H2O
Allo scopo di classificare questa reazione correttamente e stabilire un meccanismo plausibile, l'atomo di ossigeno dell'alcole è stato marcato isotopicamente come 18O (colorato in blu). Dato che questo ossigeno è ritrovato nell'estere prodotto e non nell'acqua, il gruppo idrossile dell'acido deve essere stato sostituito nella sostituzione.
Un meccanismo per questa reazione generale di esterificazione è mostrato nello schema sotto; inoltre, assieme a tale meccanismo, può essere vista una coordinata di reazione per esso.
(HO-sostituzione) CH3CO-OCH2CH3 + H2O
Allo scopo di classificare questa reazione correttamente e stabilire un meccanismo plausibile, l'atomo di ossigeno dell'alcole è stato marcato isotopicamente come 18O (colorato in blu). Dato che questo ossigeno è ritrovato nell'estere prodotto e non nell'acqua, il gruppo idrossile dell'acido deve essere stato sostituito nella sostituzione.
Un meccanismo per questa reazione generale di esterificazione è mostrato nello schema sotto; inoltre, assieme a tale meccanismo, può essere vista una coordinata di reazione per esso.
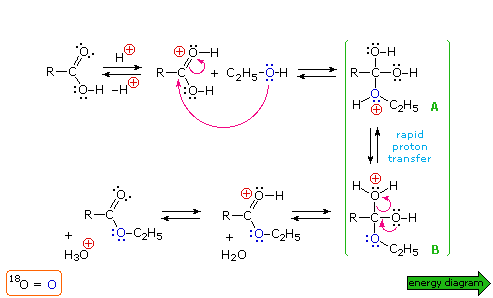
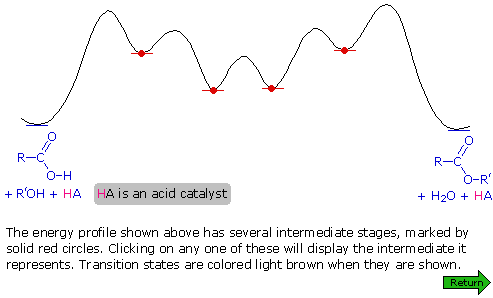 I meccanismi di addizione-eliminazione di questo tipo procedono attraverso degli intermedi tetraedrici (come A e B nello schema meccanicistico) e sono comuni nella reazione di sostituzione dell'acile. La catalisi acida è necessaria per aumentare il carattere elettrofilo dell'atomo di carbonio carbossilico, così esso si legherà più rapidamente all'ossigeno nucleofilo dell'alcole. La catalisi basica non è utile perché la base converte l'acido nella sua base coniugata anione carbossilato, una specie in cui il carattere elettrofilo del carbonio è ridotto.
Dato che un intermedio tetraedrico occupa più spazio di un gruppo carbonilico planare, dovremmo aspettarci che la velocità di questa reazione venga ritardata quando sono usati reagenti ingombrati. Per testare questa predizione l'esterificazione dell'acido acetico è stata comparata con quella dell'acido 2,2-dimetilpropanoico, (CH3)3CO2H. Qui il relativamente piccolo gruppo metile dell'acido acetico è sostituito dal più grande gruppo t-butile, e l'acido più ingombrato ha reagito cinquanta volte più lentamente dell'acido acetico. L'aumento dell'ingombro dell'alcole reagente provoca una simile riduzione della velocità.
I meccanismi di addizione-eliminazione di questo tipo procedono attraverso degli intermedi tetraedrici (come A e B nello schema meccanicistico) e sono comuni nella reazione di sostituzione dell'acile. La catalisi acida è necessaria per aumentare il carattere elettrofilo dell'atomo di carbonio carbossilico, così esso si legherà più rapidamente all'ossigeno nucleofilo dell'alcole. La catalisi basica non è utile perché la base converte l'acido nella sua base coniugata anione carbossilato, una specie in cui il carattere elettrofilo del carbonio è ridotto.
Dato che un intermedio tetraedrico occupa più spazio di un gruppo carbonilico planare, dovremmo aspettarci che la velocità di questa reazione venga ritardata quando sono usati reagenti ingombrati. Per testare questa predizione l'esterificazione dell'acido acetico è stata comparata con quella dell'acido 2,2-dimetilpropanoico, (CH3)3CO2H. Qui il relativamente piccolo gruppo metile dell'acido acetico è sostituito dal più grande gruppo t-butile, e l'acido più ingombrato ha reagito cinquanta volte più lentamente dell'acido acetico. L'aumento dell'ingombro dell'alcole reagente provoca una simile riduzione della velocità.