Fenoli
I composti in cui il gruppo idrossile è legato ad un anello aromatico sono chiamati fenoli. Il comportamento chimico dei fenoli è diverso per certi aspetti da quello degli alcoli, così è ragionevole trattarli come un gruppo simile ma caratteristicamente distinto. Una differenza simile nella reattività era stata osservata nel comparare gli alogenuri arilici con gli alogenuri alchilici. Perciò, le reazioni di sostituzione nucleofila e le reazioni di eliminazione erano comuni per gli alogenuri alchilici, ma rare con gli alogenuri arilici. Questa distinzione si estende quando si comparano alcoli e fenoli, così per tutti gli scopi pratici la sostituzione e/o l'eliminazione del gruppo idrossilico fenilico non avvengono.
1. Acidità dei fenoli
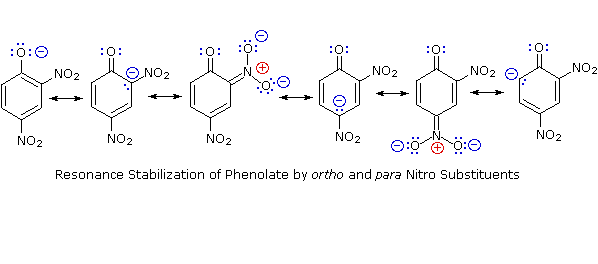
D'altra parte, la sostituzione dell'atomo di idrogeno idrossilico è ancora più facile con i fenoli, che sono circa un milione di vole più acidi degli alcoli equivalenti. Questa acidità fenolica è ulteriormente aumentata dai sostituenti elettron-attrattori in posizioni orto e para rispetto al gruppo idrossile, come mostrato nello schema seguente. L'alcole cicloesanolo è mostrato come riferimento in alto a sinistra. È degno di nota il fatto che l'influenza di un sostituente nitro è circa dieci volte più forte nella posizione para rispetto alla posizione meta, nonostante il fatto che l'ultima posizione è più vicina al gruppo idrossile. Inoltre i nitro gruppi addizionali hanno una influenza additiva se sono posizionati nelle posizioni orto o para. Il trinitro composto mostrato in basso a destra è un acido davvero forte chiamato acido picrico.
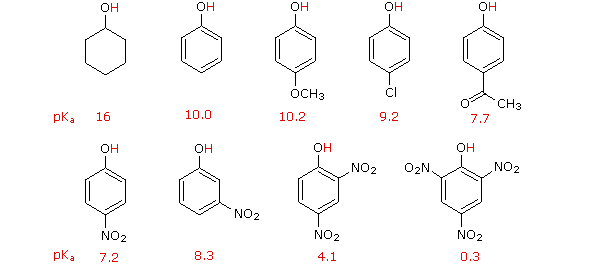
Perché il fenolo è un acido più forte del cicloesanolo? Per rispondere a questa domanda dobbiamo valutare il modo in cui un sostituente ossigenato interagisce con l'anello benzenico. Come notato nel trattamento delle reazioni di sostituzione elettrofila aromatica, precedentemente fatto, un sostituente ossigenato aumenta la reattività dell'anello e favorisce l'attacco elettrofilo nelle posizioni orto e para. È stato proposto che la delocalizzazione per risonanza di una coppia di elettroni non legata dell'ossigeno dentro il sistema ad elettroni π era responsabile di questo effetto sostituente.
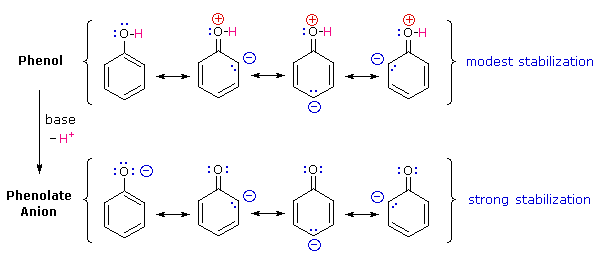
Un set simile di strutture di risonanza per questa base coniugata, l'anione fenolato, appare nelle strutture sotto.
La stabilizzazione per risonanza in questi due casi è differente. Un importante principio della risonanza è che la separazione di carica diminuisce l'importanti delle strutture canoniche che contribuiscono all'ibrido di risonanza e riduce la stabilizzazione complessiva. Le strutture che contribuiscono all'ibrido fenolo soffrono tutte di separazione di carica, che risulta dalla stabilizzazione davvero modesta di questi composto. Dall'altro lato, l'anione fenolato è già carico, e le strutture canoniche che contribuiscono alla risonanza agiscono disperdendo la carica, col risultato di una sostanziale stabilizzazione di questa specie. Le basi coniugate degli alcoli semplici non sono stabilizzate tramite delocalizzazione della carica, così l'acidità di questi composti è simile a quella dell'acqua. Un diagramma energetico che mostra l'effetto della risonanza sulle acidità del cicloesanolo e del fenolo è mostrato sotto.
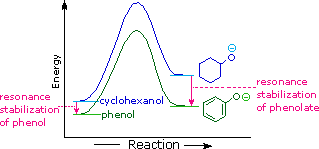
Dato che la stabilizzazione tramite risonanza della base coniugata fenolato è molto più grande della stabilizzazione del fenolo stesso, l'acidità del fenolo rispetto a quella del cicloesanolo è aumentata. La prova a supporto del fatto che la carica negativa del fenolato è delocalizzata sui carbonio orto e para dell'anello benzenico deriva dall'influenza dei sostituenti elettron-attrattori in quei siti.
2. Sostituzione dell'idrogeno idrossilico
Come con gli alcoli, l'idorgeno idrossilico fenolico è sostituito piuttosto facilmente da altri sostituenti. Per esempio, il fenolo reagisce facilmente con l'anidride acetica a dare il fenil acetate. Allo stesso modo, l'anione fenolato è un nucleofilo efficace nelle reazioni SN2, come nel secondo esempio sotto.
C6H5–OH + (CH3CO)2O  C6H5–O–COCH3 + CH3CO2H
C6H5–O–COCH3 + CH3CO2H
C6H5–O–Na+ + CH3CH2CH2–Br C6H5–O–CH2CH2CH3 + NaBr
3. Sostituzione elettrofila dell'anello aromatico fenolo
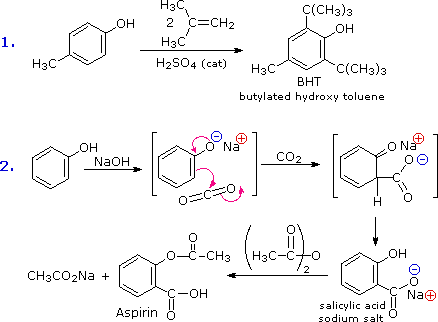
La facilità con cui l'anello aromatico dei fenoli e dei fenol eteri subisce sostituzione elettrofila è stata già notata. Due esempi sono mostrati nel seguente schema. Il primo mostra la sintesi di Friedel-Crafts del conservante alimentare BHT dal p-cresolo. La seconda reazione è interessante in quando essa dimostra ulteriormente la delocalizzazione della carica che avviene nell'anione fenolato. La CO2 è un debole elettrofilo e normalmente non reagisce con i composti aromatici; comunque, la concentrazione di carica negativa sull'anello fenolato permette la reazione di carbossilazione mostrata nel secondo passaggio. Il sale di sodio dell'acido salicilico è il prodotto maggioritario, e la preferenza per la posizione orto può riflettere l'influenza del catione sodio. Questa reazione è chiama reazione di Kolbe-Schmidt, ed è servita nella preparazione dell'aspirina, come l'ultimo passaggio illustra.
4. Ossidazione dei fenoli
I fenoli sono piuttosto facilmente ossidati nonostante l'assenza di un atomo di idrogeno sul carbonio che porta l'idrossile. Tra i prodotti colorati derivati dall'ossidazione del fenolo con acido cromico c'è il composto dicarbonilico p-benzochinone (anche noto come 1,4-benzochinone o semplicemente chinone); è anche noto l'isomero orto. Questi composti sono facilmente ridotti nei loro analoghi diidrossibenzeni, ed è da questi composti che sono meglio preparati i chinoni. Notate che non esistono m-chinoni aventi strutture simili. Gli equilibri redox tra gli stati di ossidazione dei diidrossibenzeni idrochinone e catecolo e dei loro chinoni sono così facili che sono generalmente preferiti ossidanti più blandi del cromato. Un tale ossidante è il sale di Fremy, mostrato sotto. Agenti riducenti diversi dal cloruro di stagno(II) (ad esempio NaBH4) possono essere usati per le reazioni inverse.
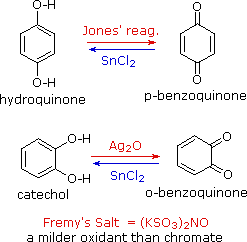
La posizione dell'equilibrio redox chinone-idrossichinone è proporzionale alla radice quadrata della concentrazione dello ione idrogeno, come mostrato dalle seguenti semi-reazioni (gli elettroni sono colorati in blu). Il potenziale d'elettrodo per questa interconversione può inoltre essere usato per misurare il pH delle soluzioni.
2e–
Chinone + 2H+  Idrochinone
Idrochinone
–2e–
Sebbene l'ossidazione con acido cromico dei fenoli aventi una posizione para non sostituita dia un po' di p-chinone come prodotto, la reazione è complessa e non è sinteticamente utile.
È stato scoperto che la salcomina, un complesso di cobalto(II), lega l'ossigeno reversibilmente in soluzione, e catalizza l'ossidazione di diversi fenoli sostituiti nei corrispondenti p-chinoni. La struttura della salcomina ed un esempio di questa reazione sono mostrati nella seguente equazione. Il solvente di scelta per queste ossidazioni è di solito il metanolo o la dimetilformammide (DMF).
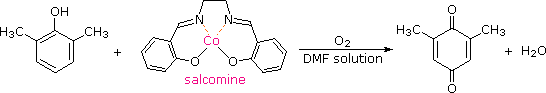
 La preparazione del t-butil ipoclorito dall'alcole t-butilico è un esempio di alogenazione elettrofila dell'ossigeno, ma questa reazione è ristretta agli alcoli terziari perché gli ipocloriti primarie e secondari perdono HCl a dare aldeidi e chetonii. Nella seguente equazione l'elettrofilo può essere considerato come Cl+.
(CH3)3C–O–H + Cl2 + NaOH
La preparazione del t-butil ipoclorito dall'alcole t-butilico è un esempio di alogenazione elettrofila dell'ossigeno, ma questa reazione è ristretta agli alcoli terziari perché gli ipocloriti primarie e secondari perdono HCl a dare aldeidi e chetonii. Nella seguente equazione l'elettrofilo può essere considerato come Cl+.
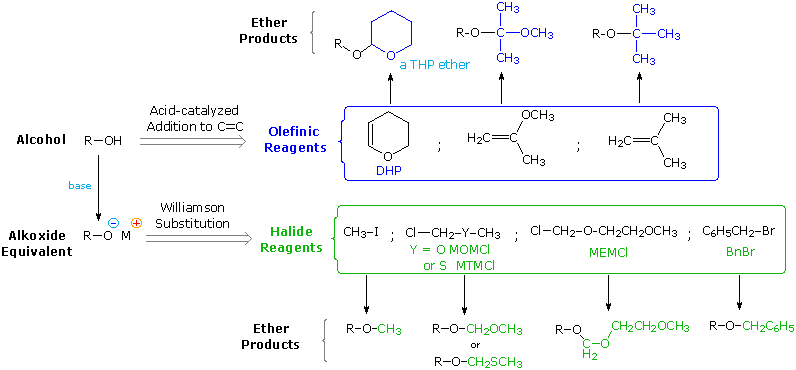
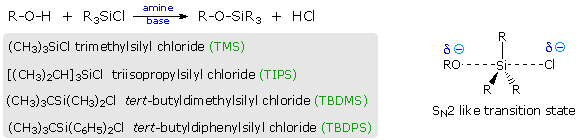
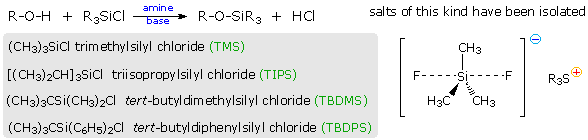
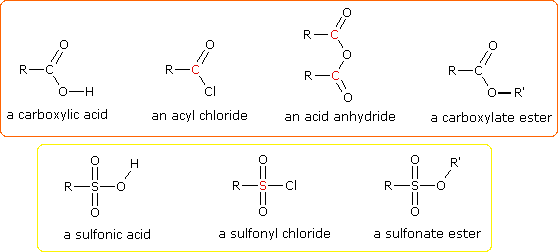
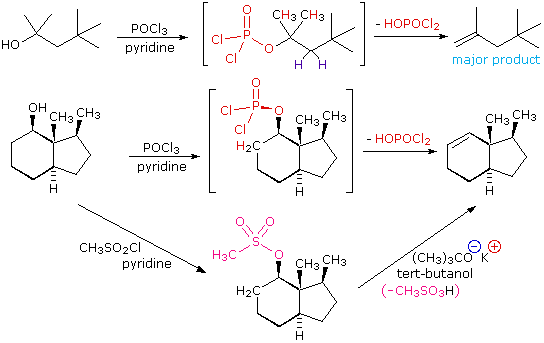
(CH3)3C–O–H + Cl2 + NaOH 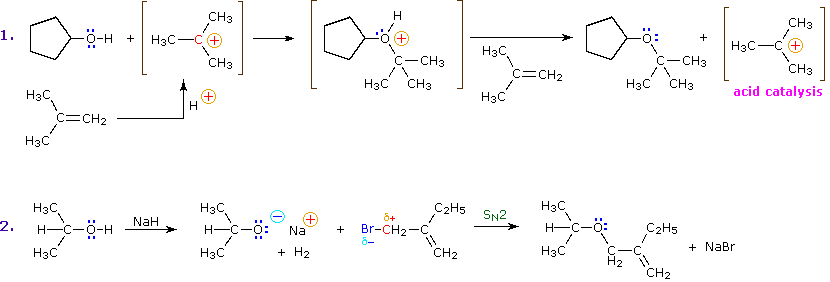 Una delle più importanti reazioni di sostituzione all'ossigeno è la formazione di un estere che risulta dalla reazione degli alcoli con i derivati elettrofili degli acidi carbossilici e solfonici. La seguente illustrazione mostra le formule generali di questi reagenti ed i loro prodotti (esteri), in cui il gruppo R'–O– rappresenta la parte alcolica. L'atomo elettrofilo nei cloruri acilici e nelle anidridi è colorato in rosso.
Una delle più importanti reazioni di sostituzione all'ossigeno è la formazione di un estere che risulta dalla reazione degli alcoli con i derivati elettrofili degli acidi carbossilici e solfonici. La seguente illustrazione mostra le formule generali di questi reagenti ed i loro prodotti (esteri), in cui il gruppo R'–O– rappresenta la parte alcolica. L'atomo elettrofilo nei cloruri acilici e nelle anidridi è colorato in rosso.
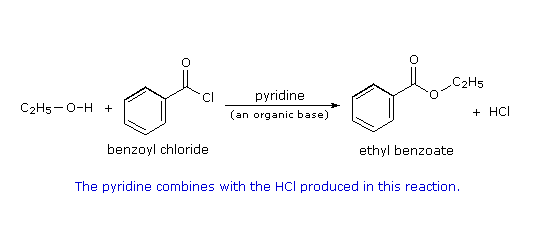
 Esterificazione di un cloruro acilico:
Esterificazione di un cloruro acilico:
 Esterificazione di un'anidride:
Esterificazione di un'anidride:
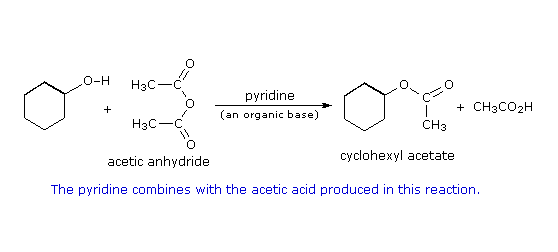 Formazione del mesilato:
Formazione del mesilato:
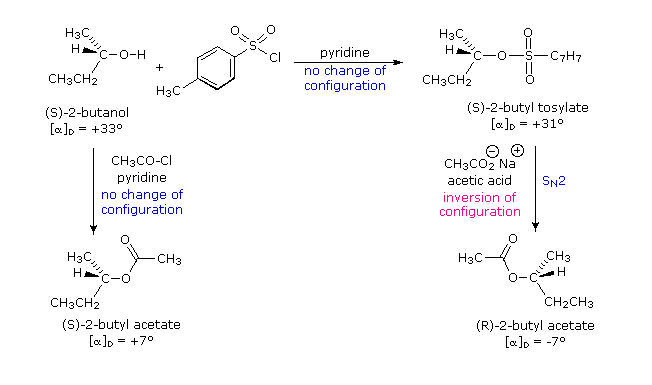
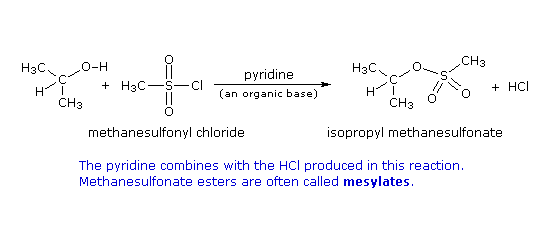 Formazione del tosilato:
Formazione del tosilato:
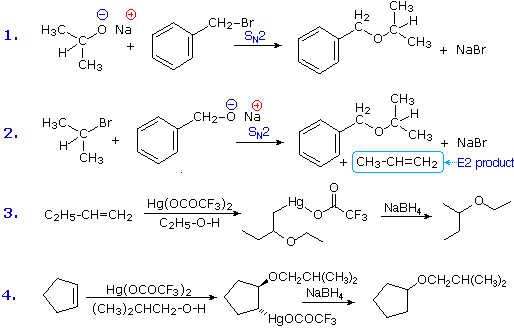
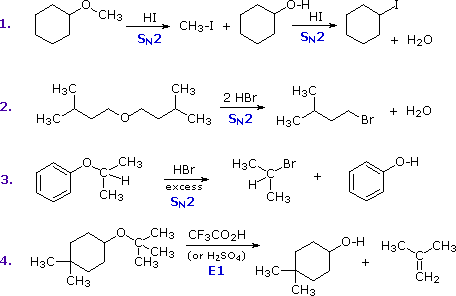
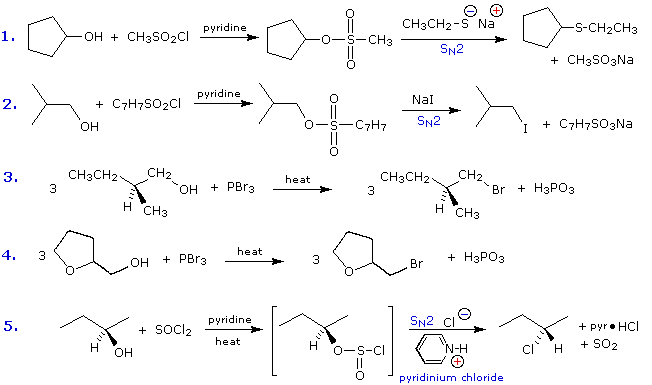
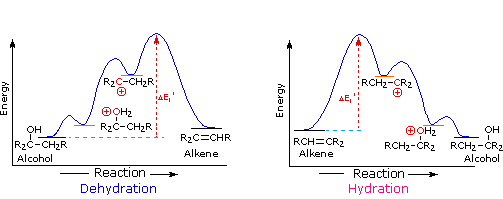
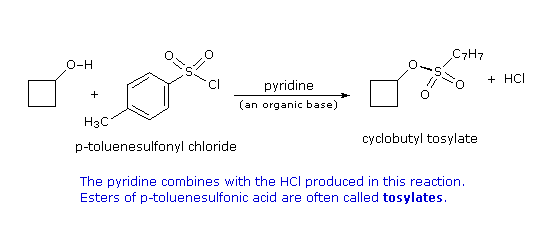 2. Sostituzione nucleofila del gruppo idrossile
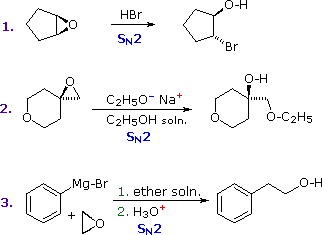
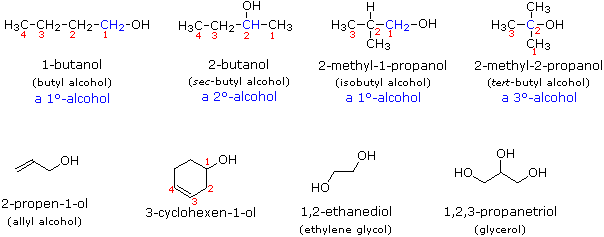
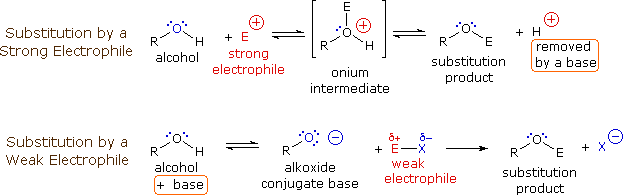
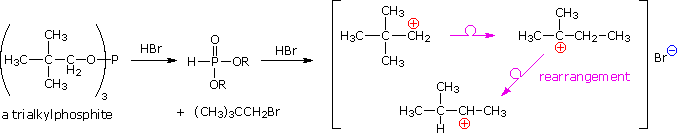
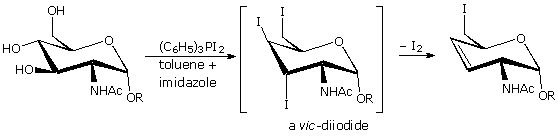
Usando il comportamento chimico degli alogenuri alchilici come riferimento, siamo stimolati a guardare le analoghe reazioni di sostituzione ed eliminazione degli alcoli. La differenza principale, naturalmente, è un cambiamento nel gruppo uscente dall'alogenuro all'idrossido. Dato che l'ossigeno è leggermente più elettronegativo del cloro (3.5 vs. 2.8 secondo Pauling), ci aspettiamo che il legame C-O sia più polare del legame C-Cl. Inoltre, una misura indipendente del carattere elettrofilo degli atomi di carboni dai loro chemical shifts all'NMR (sia 13C sia protoni α), indica che i sostituenti ossigeno e cloro esercitano una influenza elettron attrattrice simile quando legati ad atomi di carbonio ibridati sp3. Malgrado questa prova di fondo evidente, gli alcoli non subiscono le stesse reazioni SN2 comunemente osservate con gli alogenuri alchilici. Per esempio, la rapida reazione SN2 dell'1-bromobutano con sodio cianuro, mostrata sotto, non ha analogo quando l'1-butanolo viene trattato con sodio cianuro. Infatti l'alcole etilico è spesso usato come solvente per le reazioni di sostituzione di alogenuri alchilici come questa.
CH3CH2CH2CH2–Br + Na+CN–
2. Sostituzione nucleofila del gruppo idrossile
Usando il comportamento chimico degli alogenuri alchilici come riferimento, siamo stimolati a guardare le analoghe reazioni di sostituzione ed eliminazione degli alcoli. La differenza principale, naturalmente, è un cambiamento nel gruppo uscente dall'alogenuro all'idrossido. Dato che l'ossigeno è leggermente più elettronegativo del cloro (3.5 vs. 2.8 secondo Pauling), ci aspettiamo che il legame C-O sia più polare del legame C-Cl. Inoltre, una misura indipendente del carattere elettrofilo degli atomi di carboni dai loro chemical shifts all'NMR (sia 13C sia protoni α), indica che i sostituenti ossigeno e cloro esercitano una influenza elettron attrattrice simile quando legati ad atomi di carbonio ibridati sp3. Malgrado questa prova di fondo evidente, gli alcoli non subiscono le stesse reazioni SN2 comunemente osservate con gli alogenuri alchilici. Per esempio, la rapida reazione SN2 dell'1-bromobutano con sodio cianuro, mostrata sotto, non ha analogo quando l'1-butanolo viene trattato con sodio cianuro. Infatti l'alcole etilico è spesso usato come solvente per le reazioni di sostituzione di alogenuri alchilici come questa.
CH3CH2CH2CH2–Br + Na+CN– 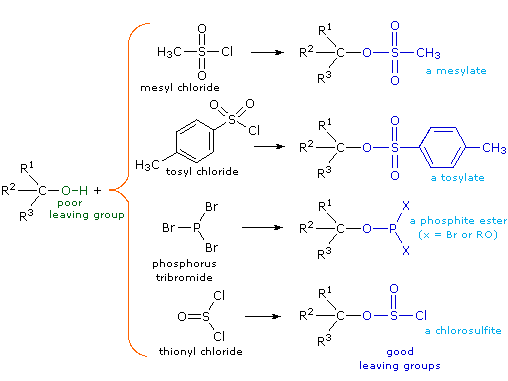
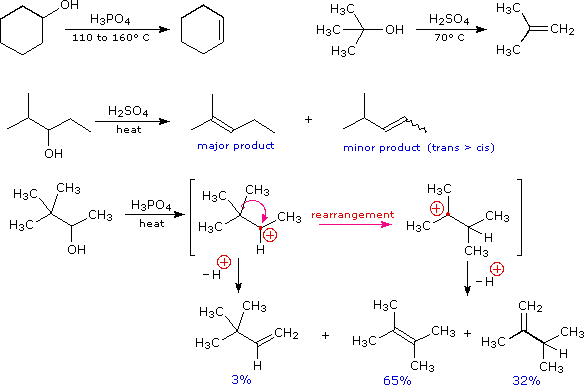
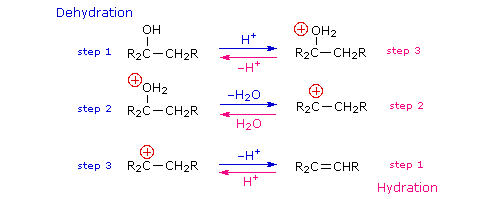
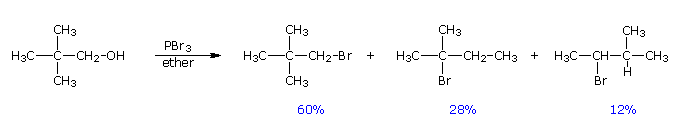
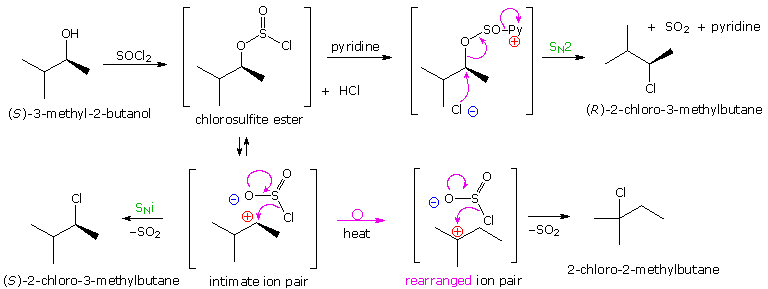
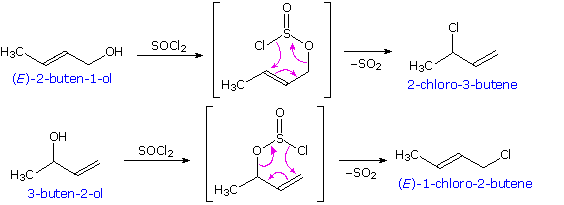
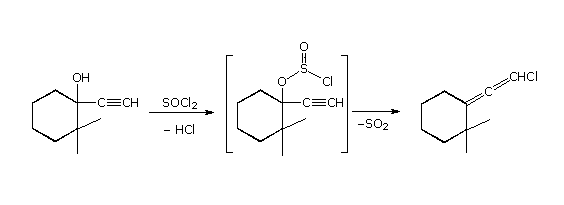
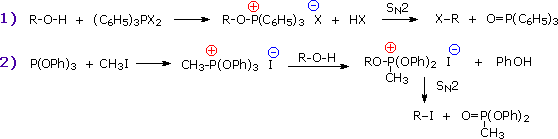
 I meccanismi di reazioni per queste trasformazioni sono mostrati sotto. Per le prime due reazioni il diagramma del meccanismo mostra anche gli stati di ossidazione del carbonio (numeri arabi in blu) e del cromo (numeri romani). La base generale, B:, usata in questi meccanismi può essere una qualsiasi dall'acqua alla piridina, a seconda della reazione specifica.
I meccanismi di reazioni per queste trasformazioni sono mostrati sotto. Per le prime due reazioni il diagramma del meccanismo mostra anche gli stati di ossidazione del carbonio (numeri arabi in blu) e del cromo (numeri romani). La base generale, B:, usata in questi meccanismi può essere una qualsiasi dall'acqua alla piridina, a seconda della reazione specifica.
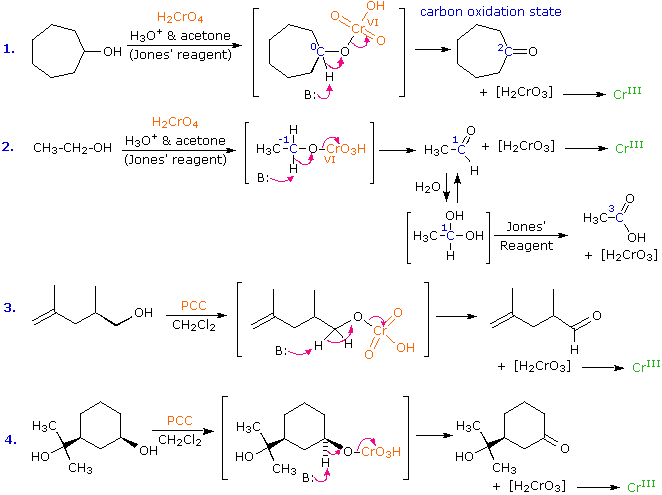 Due requisiti strutturali per l'ossidazione a composti carbonilici dovrebbero essere ora ovvi:
1. L'atomo di carbonio legato all'ossigeno deve anche portare un atomo di idrogeno.
Gli alcoli terziari (R3C–OH) non possono essere ossidati in questa maniera.
2. L'atomo di ossigeno deve essere legato ad un atomo di idrogeno così che un intermedio estere cromato (o un altro gruppo uscente idoneo) possa essere formato.
Gli eteri (R–O–R) non possono essere ossidati in questa maniera.
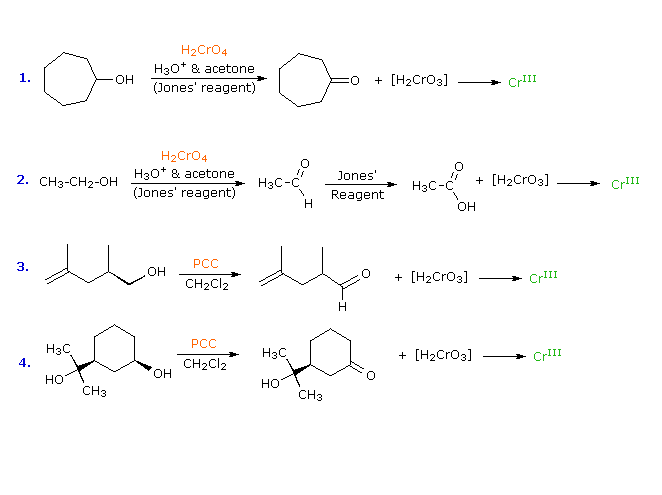
La quarta reazione sopra illustra il fallimento da parte degli alcoli terziari di subire ossidazione. La seconda reazione spiega perché gli alcoli primari subiscono ulteriore ossidazione da parte del reagente di Jones. Il sistema solvente acquoso usato con questo reagente permette l'idratazione (addizione di acqua) al gruppo carbonilico dell'aldeide. L'idrato risultante (struttura mostrata sotto l'aldeide) rispetta entrambi i requisiti affermati sopra, e viene ulteriomente ossidato tramite lo stesso meccanismo. L'acqua non è presente quando viene usato il reagente PCC, così l'ossidazione si ferma allo stadio di aldeide.
Un altro agente ossidante contenente cromato, simile al PCC, è il piridinio dicromato, (C5H5NH+)2Cr2O7–2, noto tramite l'acronimo PDC. Sia il PCC che il PDC sono solidi cristallini arancioni che sono solubili in molti solventi organici. Dato che il PDC è meno acido del PCC è spesso usato per ossidare alcoli che possono essere sensibili agli acidi. In soluzione di metilene cloruro, il PDC ossida gli alcoli primari e secondari in circa lo stesso modo del PCC, ma molto più lentamente. Comunque, in soluzione satura di DMF gli alcoli primari sono ossidati ad acidi carbossilici. In entrambi i solventi gli alcoli allilici sono ossidati efficacemente ad enali ed enoni coniugati rispettivamente.
Dei metodi di ossidazione degli alcoli ne ho già parlato qui
Due requisiti strutturali per l'ossidazione a composti carbonilici dovrebbero essere ora ovvi:
1. L'atomo di carbonio legato all'ossigeno deve anche portare un atomo di idrogeno.
Gli alcoli terziari (R3C–OH) non possono essere ossidati in questa maniera.
2. L'atomo di ossigeno deve essere legato ad un atomo di idrogeno così che un intermedio estere cromato (o un altro gruppo uscente idoneo) possa essere formato.
Gli eteri (R–O–R) non possono essere ossidati in questa maniera.
La quarta reazione sopra illustra il fallimento da parte degli alcoli terziari di subire ossidazione. La seconda reazione spiega perché gli alcoli primari subiscono ulteriore ossidazione da parte del reagente di Jones. Il sistema solvente acquoso usato con questo reagente permette l'idratazione (addizione di acqua) al gruppo carbonilico dell'aldeide. L'idrato risultante (struttura mostrata sotto l'aldeide) rispetta entrambi i requisiti affermati sopra, e viene ulteriomente ossidato tramite lo stesso meccanismo. L'acqua non è presente quando viene usato il reagente PCC, così l'ossidazione si ferma allo stadio di aldeide.
Un altro agente ossidante contenente cromato, simile al PCC, è il piridinio dicromato, (C5H5NH+)2Cr2O7–2, noto tramite l'acronimo PDC. Sia il PCC che il PDC sono solidi cristallini arancioni che sono solubili in molti solventi organici. Dato che il PDC è meno acido del PCC è spesso usato per ossidare alcoli che possono essere sensibili agli acidi. In soluzione di metilene cloruro, il PDC ossida gli alcoli primari e secondari in circa lo stesso modo del PCC, ma molto più lentamente. Comunque, in soluzione satura di DMF gli alcoli primari sono ossidati ad acidi carbossilici. In entrambi i solventi gli alcoli allilici sono ossidati efficacemente ad enali ed enoni coniugati rispettivamente.
Dei metodi di ossidazione degli alcoli ne ho già parlato qui 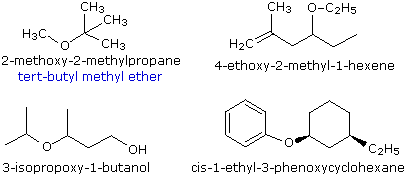
 sono chiamati disolfuri. Per esempio, (CH3)3C–S–CH3 è il t-butil metil solfuro. I solfuri sono chimicamente più reattivi degli eteri, riflettendo la maggiore nucleofilicità dello zolfo rispetto all'ossigeno.
sono chiamati disolfuri. Per esempio, (CH3)3C–S–CH3 è il t-butil metil solfuro. I solfuri sono chimicamente più reattivi degli eteri, riflettendo la maggiore nucleofilicità dello zolfo rispetto all'ossigeno.