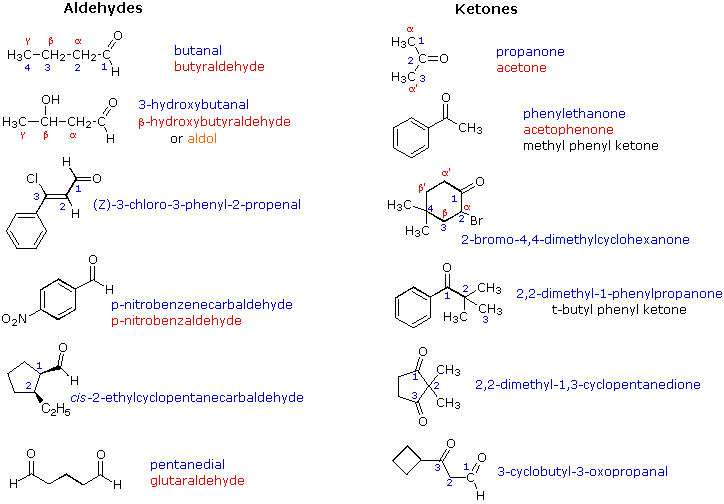
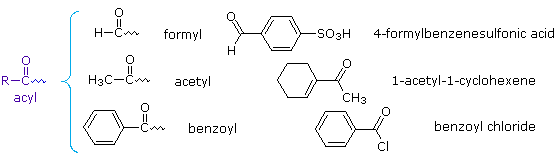
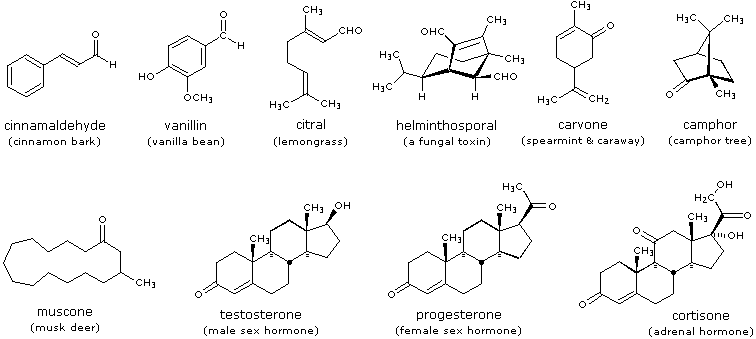
Reazioni di aldeidi e chetoni
1. Reazioni di addizione reversibili
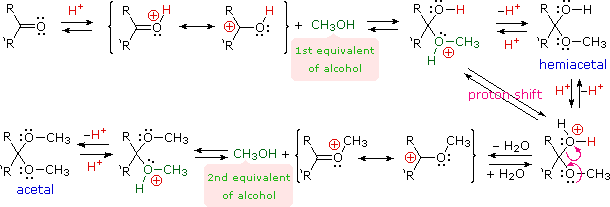
A. Idratazione e formazione di emiacetali
È stato dimostrato (sopra) che l'acqua si addiziona rapidamente alla funzione carbonilica di aldeidi e chetoni. Nella maggior parte dei casi il risultante idrato (un diolo geminale) è instabile rispetto ai reagenti e non può essere isolato. Eccezioni a questa regola esistono, ed una di queste è ad esempio il caso della formaldeide (vorrei ricordare che essa è un gas nel suo stato monomerico puro!). Qui la più debole compenente π del doppio legame carbonilico, rispetto ad altre aldeidi o chetoni, e la piccola dimensione dei sostituenti idrogeno favorisce l'addizione. Perciò, una soluzione di formaldeide in acqua (formalina) è quasi esclusivamente l'idrato, o polimeri dell'idrato. Simili addizioni reversibili di alcoli ad aldeidi e chetoni avvengono. Il prodotti di addizione parimenti instabili sono chiamati emiacetali.
R2C=O + R'OH  R'O–(R2)C–O–H (un emiacetale)
R'O–(R2)C–O–H (un emiacetale)
B. Formazione di acetali
Gli acetali sono i derivati dieteri geminali di aldeidi e chetoni, formati tramite reazione con due equivalenti di un alcole ed eliminazione di acqua. I derivati chetonici di questo genere erano una volta chiamati chetali, ma l'uso moderno ha rigettato questo termine. La seguente equazione mostra il cambiamento stechiometrico complessivo nella formazione degli acetali, ma viene utilizzata una freccia tratteggiata perché questa conversione non avviene per semplice miscelazione dei reagenti.
R2C=O + 2 R'OH  R2C(OR')2 + H2O (un acetale)
R2C(OR')2 + H2O (un acetale)
Al fine di ottenere un efficace formazione dell'acetale devono essere migliorate due caratteristiche addizionali. Primo, un catalizzatore acido deve essere usato; e secondo, l'acqua prodotta dall'acetale deve essere rimossa dalla reazione. L'ultima caratteristica è importante, dato che la formazione dell'acetale è reversibile. Inoltre, una volta che sono ottenuti gli acetali puri essi possono essere idrolizzati di nuovo nei loro componenti di partenza per trattamento con acidi acquosi. Il meccanismo mostrato qui si applica sia alla formazione dell'acetale sia all'idrolisi dell'acetale tramite il principio di reversibilità microscopica.
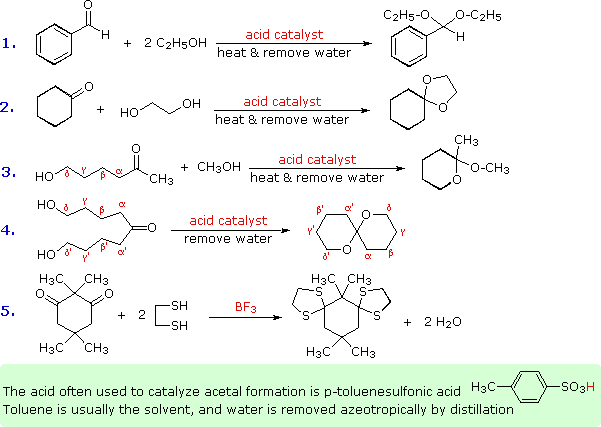
Alcuni esempi di formazione di acetali sono presentati nel seguente schema. Come notato, l'acido p-toluenesolfonico (pKa = -2) è spesso il catalizzatore per tali reazioni. Sono necessari due equivalenti dell'alcole reagente, ma questi possono essere forniti da un equivalente di un diolo (esempio #2). Il coinvolgimento intramolecolare di un gruppo γ- o δ-idrossile (come negli esempi #3 e 4) può avvenire, ed è spesso più facile rispetto a quanto avviene nella reazione intermolecolare. I tioli danno i tioacetali (esempio #5). In questo caso le funzioni carboniliche sono relativamente ingombrate, ma usando un eccesso di etanditiolo come solvente e l'acido di Lewis BF3 come catalizzatore si ottiene una buona resa del bis-tioacetale. I tioacetali sono generalmente più difficili da idrolizzare rispetto agli acetali.
L'importanza degli acetali come derivati carbonilici risiede principalmente nella loro stabilità e mancanza di reattività in ambienti da neutri a fortemente basici. Finché essi non vengono trattati con acidi, specialmente acidi acquosi, gli acetali esibiscono tutti la mancanza di reattività associata agli eteri in generale.
Tra le più utili e caratteristiche reazioni di aldeidi e chetoni c'è la loro reattività verso i metallo idruri, reagenti alchilici ed arilici fortemente nucleofili (e basici) (discussi a breve). Se il gruppo funzionale carbonile viene convertito in un acetale questi potenti reagenti non hanno effetto; quindi, gli acetali sono eccellenti gruppi protettivi, quando queste reazioni di addizione irreversibili devono essere prevenute.
C. Formazione di immine e composti correlati
La reazioni di aldeidi e chetoni con ammoniaca o amine primare forma i derivati imminici o immine, anche noti come basi di Schiff, (composti aventi una funzione C=N). Questa reazione gioca un importante ruolo nella sintesi di amine secondarie, come discusso in precedenza. In questa reazione, che è acido-catalizzata e reversibile nel senso medesiamo della formazione dell'acetale, viene persa acqua.
R2C=O + R'NH2  R'NH–(R2)C–O–H R2C=NR' + H2O
R'NH–(R2)C–O–H R2C=NR' + H2O
È stato proposto un meccanismo di addizione-eliminazione per questa reazione, ed un'animazione che mostra questo meccanismo è mostrata sotto.
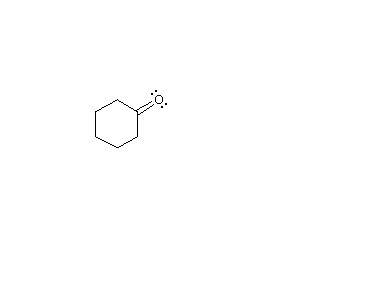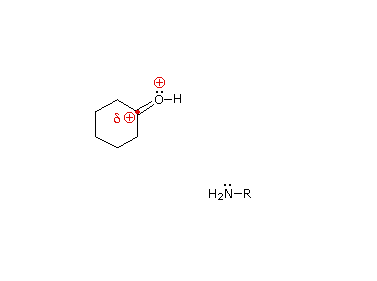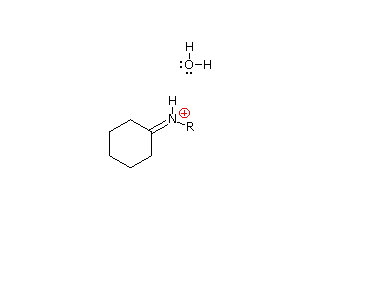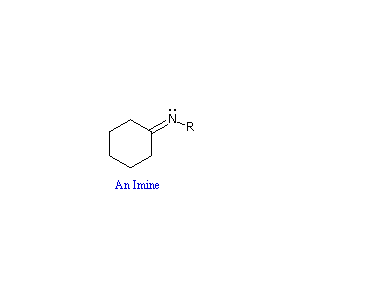
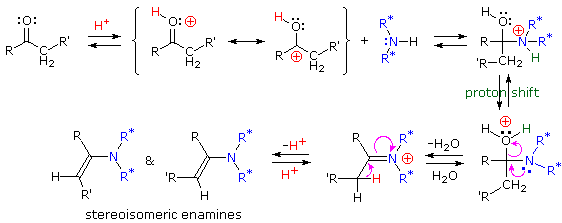
Le immine sono qualche volta difficili da isolare e purificare a causa della loro sensibilità all'idrolisi. Di conseguenza, altri reagenti del tipo Y–NH2 sono stati studiati, ed è stato trovato che danno prodotti stabili (R2C=N–Y) utili nel caratterizzare le aldeidi ed i chetoni da cui sono preparati. Alcuni di questi reagenti sono elencati nella tabella seguente, assieme alle strutture e ai nomi dei loro prodotti della reazione con il carbonile. Un aspetto interessante di questi derivati carbonilici è che sono possibili degli stereoisomeri quando i gruppi R del reagente carbonilico sono diversi. Perciò, la benzaldeide forma due ossime stereoisomeriche, un isomero bassofondente, avente il gruppo idrossile cis rispetto all'idrogeno dell'aldeide (chiamato syn), ed un isomero più altobollente in cui il gruppo idrossile e l'idrogeno sono trans (l'isomero anti). A temperatura ambiente o sotto la configurazione dell'atomo di idrogeno doppiamente legato è apparentemente fissata in una forma trigonale, nonostante le configurazioni piramidali, che interconvertono rapidamente, delle amine ibridizzate sp3.
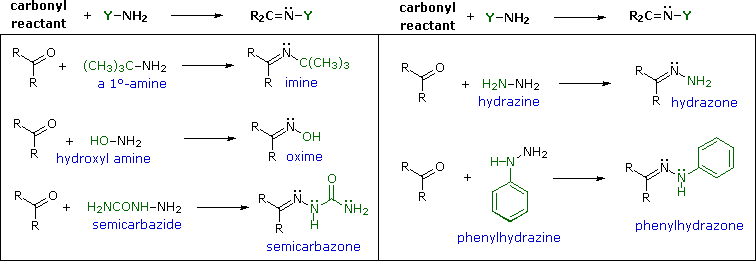
Con l'eccezione degli idrazoni non sostituiti, questi derivati sono facilmente preparati e sono spesso solidi cristallini - anche quando l'aldeide o il chetone genitore è un liquido. Dato che i m.p. possono essere determinati più rapidamente dei b.p., i derivati come questi sono utili per il paragone e l'indentificazione dei composti carbonilici. Se l'anello aromatico della fenilidrazina è sostituito con nitro gruppo nelle posizioni 2 & 4, il reagente risultante e gli idrazoni derivati che esso dà sono fortemente colorati, rendendoli facili da identificare. Bisogna notare che nonostante la semicarbazide abbia due gruppi amino (–NH2) solo uno di essi è un'amina reattiva. L'altro è tipo ammide ed è disattivato dal gruppo carbonilico adiacente.
La velocità a cui questi composti tipo immina sono formati è generalmente più grande vicino ad un pH di 5, e cade a pH più alti e più bassi. Questo è d'accordo con una generale catalisi acida in cui l'acido coniugato del reagente carbonilico si combina con un gruppo amino libero, come mostrato nella animazione sopra. Ad un pH alto ci sarà una bassa concentrazione che sta scomparendo dell'acido coniugato del carbonile, e ad un pH basso la maggior parte dell'amina reagente sarà legata come suo acido coniugato ammonio. Con l'eccezione della formazione dell'immina stessa, la maggiro parte di queste reazioni di derivatizzazione non richiedono la rimozione attiva di acqua (non mostrata come prodotto nelle equazioni precedenti). Le reazioni sono reversibili, ma l'equilibrio non è stabilito instantaneamente ed i prodotti spesso precipitano dalla soluzione non appena si formano.
D. Formazione dell'enammina
Le reazioni precedenti hanno tutte coinvolto reagenti del tipo: Y–NH
2, cioè reazioni con un gruppo amino primario. La maggior parte delle aldeidi e dei chetoni reagisce anche con le amine secondarie a dar prodotti noti come
enammine. Due esempi di queste reazioni sono presentati nel seguente schema. Bisogna notare che, come la formazione di acetali, queste sono reazioni reversibili acido-catalizzate in cui l'acqua viene persa. Di conseguenza, le enammine sono facilmente riconvertite nei loro composti carbonilici tramite idrolisi acido-catalizzata.
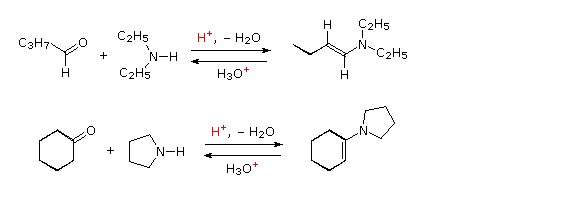
Sotto viene mostrato un meccanismo per la formazione dell'enammina.
E. Formazione della cianoidrina
L'ultimo esempi di addizione reversibile è quella del cianuro di idrogeno (HC≡N), che si addiziona alle aldeidi e a molti chetoni a dare prodotti noti come
cianoidrine.
RCH=O + H–C≡N
RCH(OH)CN (una cianoidrina)
Dato che il cianuro di idrogeno stesso è un acido (pK
a = 9.25), l'addizione non è acido-catalizzata. Infatti, per risultati migliori l'anione cianuro, C≡N
–, deve essere presente, il che significa che la base deve essere aggiunta in quantità catalitica. La formazione della cianoidrina è debolmente esotermica, ed è favorita per le aldeidi, e per i chetoni ciclici non ingombrati e per i metil chetoni. Due esempi di tali reazioni sono mostrati sotto.
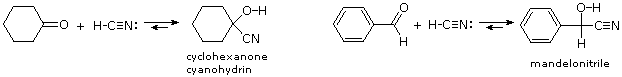
La cianoidrina dalla benzaldeide è chiamata mandelonitrile. La reversibilità della formazione della cianoidrina è messa in atto dal millepiede
Apheloria corrugata in un notevole meccanismo di difesa. Questo artropode rilascia il mandelonitrile da una ghiandola di immagazzinamento interna verso una camera esterna, dove è enzimaticamente rotto di nuovo a benzaldeide e cianuro di idrogeno prima di essere spruzzato ad un nemico.
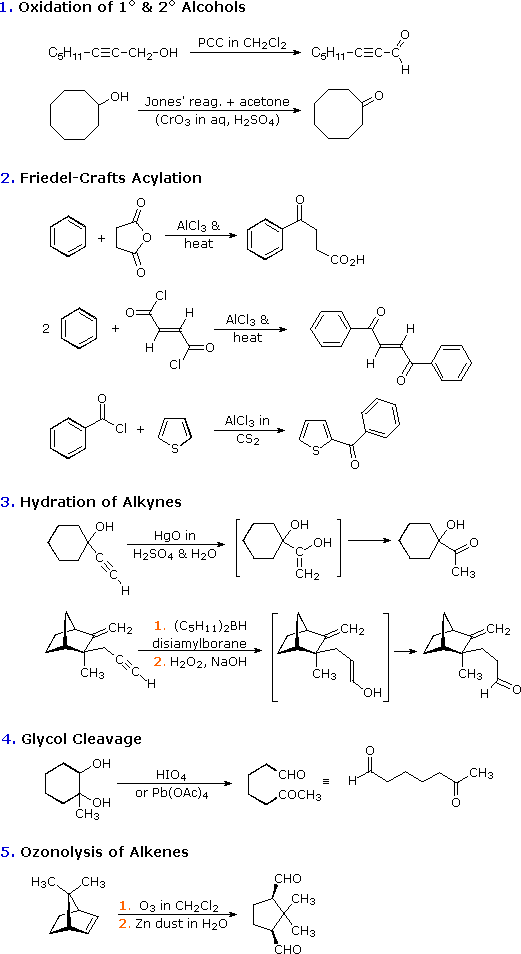 Con l'eccezione della acilazione di Friedel-Crafts, questi metodi non aumentano la dimensione o la complessità delle molecole. Nelle sezioni seguenti di questo capitolo scopriremo che una delle più utili caratteristiche delle aldeidi e dei chetoni è la loro reattività verso i nucleofili al carbonio, e la risultante elaborazione della struttura molecolare che risulta. In breve, le aldeidi ed i chetoni sono intermedi importanti per l'assemblaggio o sintesi di molecole organiche complesse.
4. Proprietà di aldeidi e chetoni
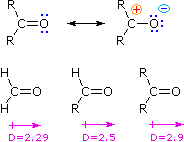
Un paragone tra le proprietà e la reattività di aldeidi e chetoni e quelle degli alcheni è giustificato, dato che entrambi hanno come gruppo funzionale un doppio legame. A causa della maggiore elettronegatività dell'ossigeno, il gruppo carbonile è polare, e le aldeidi ed i chetoni hanno momenti dipolari molecolari maggiori (D) rispetto a quanto fanno gli alcheni. Le strutture di risonanza sotto illustrano questa polarità, ed i momenti dipolari relativi della formaldeide, di altre aldeidi e chetoni confermano l'influenza stabilizzante che i sostituenti alchilici hanno sui carbocationi (maggiore il momento dipolare maggiore il carattere polare del gruppo carbonilico).
Con l'eccezione della acilazione di Friedel-Crafts, questi metodi non aumentano la dimensione o la complessità delle molecole. Nelle sezioni seguenti di questo capitolo scopriremo che una delle più utili caratteristiche delle aldeidi e dei chetoni è la loro reattività verso i nucleofili al carbonio, e la risultante elaborazione della struttura molecolare che risulta. In breve, le aldeidi ed i chetoni sono intermedi importanti per l'assemblaggio o sintesi di molecole organiche complesse.
4. Proprietà di aldeidi e chetoni
Un paragone tra le proprietà e la reattività di aldeidi e chetoni e quelle degli alcheni è giustificato, dato che entrambi hanno come gruppo funzionale un doppio legame. A causa della maggiore elettronegatività dell'ossigeno, il gruppo carbonile è polare, e le aldeidi ed i chetoni hanno momenti dipolari molecolari maggiori (D) rispetto a quanto fanno gli alcheni. Le strutture di risonanza sotto illustrano questa polarità, ed i momenti dipolari relativi della formaldeide, di altre aldeidi e chetoni confermano l'influenza stabilizzante che i sostituenti alchilici hanno sui carbocationi (maggiore il momento dipolare maggiore il carattere polare del gruppo carbonilico).
 Ci aspettiamo, quindi, che le aldeidi ed i chetoni avranno maggiori punti di ebollizione rispetto ad alcheni di simili dimensioni. Inoltre, la presenza dell'ossigeno con i suoi doppietti elettronici di non legame rende le aldeidi ed i chetoni accettori di legami ad idrogeno, ed aumenterebbe la loro solubilità in acqua rispetto agli idrocarburi. Esempi specifici di queste relazioni sono fornite sotto.
(CH3)2C=CH2 PM:56 b.p.:-7.0 ºC Solubilità in acqua: 0.04 g/100
(CH3)2C=O PM:58 b.p.: 56.5 ºC Solubilità in acqua: infinita
CH3CH2CH2CH=CH2 PM: 70 b.p.: 30.0 ºC Solubilità in acqua: 0.03 g/100
CH3CH2CH2CH=O PM: 72 b.p.: 76.0 ºC Solubilità in acqua: 7 g/100
Ci aspettiamo, quindi, che le aldeidi ed i chetoni avranno maggiori punti di ebollizione rispetto ad alcheni di simili dimensioni. Inoltre, la presenza dell'ossigeno con i suoi doppietti elettronici di non legame rende le aldeidi ed i chetoni accettori di legami ad idrogeno, ed aumenterebbe la loro solubilità in acqua rispetto agli idrocarburi. Esempi specifici di queste relazioni sono fornite sotto.
(CH3)2C=CH2 PM:56 b.p.:-7.0 ºC Solubilità in acqua: 0.04 g/100
(CH3)2C=O PM:58 b.p.: 56.5 ºC Solubilità in acqua: infinita
CH3CH2CH2CH=CH2 PM: 70 b.p.: 30.0 ºC Solubilità in acqua: 0.03 g/100
CH3CH2CH2CH=O PM: 72 b.p.: 76.0 ºC Solubilità in acqua: 7 g/100
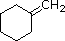 PM: 96 b.p.: 103.0 ºC Solubilità in acqua: insolubile
PM: 96 b.p.: 103.0 ºC Solubilità in acqua: insolubile
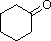 PM: 98 b.p.: 155.6 ºC Solubilità in acqua: 5 g/100
La polarità del gruppo carbonile ha anche un profondo effetto sulla sua reattività chimica, comparata con quella dei doppi legami non polari degli alcheni. Quindi, l'addizione reversibile di acqua alla funzione carbonile è veloce, mentre l'addizione di acqua agli alcheni è incommensurabilmene lenta in assenza di un catalizzatore acido forte. Curiosamente, le energie relative di legame influenzano le termodinamiche di tali reazioni di addizione nel senso opposto.
Il C=C degli alcheni ha un'energia di legame media di 146 kcal/mole. Dato che un legame σ C–C ha una energia di legame di 83 kcal/mole, l'energia del legame π può essere stimata a 63 kcal/mole (cioè men o dell'energia del legame σ). L'energia del legame C=O di un gruppo carbonile, d'altra parte, varia con la sua posizione, come segue:
H2C=O 170 kcal/mole
RCH=O 175 kcal/mole
R2C=O 180 kcal/mole
Il legame σ C–O si è scoperto avere una energia di legame media di 86 kcal/mole. Di conseguenza, con l'eccezione della formaldeide, la funzione carbonilica di aldeidi e chetoni ha una energia di legame π maggiore di quella del legame σ, in contrato con la relazione π-σ nel legame C=C. Questo suggerisce che le reazioni di addizione ai gruppi carbonile dovrebbero essere termodinamicamente sfavoriti, come nel caso dell'addizione di acqua. Tutto questo è riassunto nel seguente schema (i valori di ΔH° sono per la reazione di addizione).
PM: 98 b.p.: 155.6 ºC Solubilità in acqua: 5 g/100
La polarità del gruppo carbonile ha anche un profondo effetto sulla sua reattività chimica, comparata con quella dei doppi legami non polari degli alcheni. Quindi, l'addizione reversibile di acqua alla funzione carbonile è veloce, mentre l'addizione di acqua agli alcheni è incommensurabilmene lenta in assenza di un catalizzatore acido forte. Curiosamente, le energie relative di legame influenzano le termodinamiche di tali reazioni di addizione nel senso opposto.
Il C=C degli alcheni ha un'energia di legame media di 146 kcal/mole. Dato che un legame σ C–C ha una energia di legame di 83 kcal/mole, l'energia del legame π può essere stimata a 63 kcal/mole (cioè men o dell'energia del legame σ). L'energia del legame C=O di un gruppo carbonile, d'altra parte, varia con la sua posizione, come segue:
H2C=O 170 kcal/mole
RCH=O 175 kcal/mole
R2C=O 180 kcal/mole
Il legame σ C–O si è scoperto avere una energia di legame media di 86 kcal/mole. Di conseguenza, con l'eccezione della formaldeide, la funzione carbonilica di aldeidi e chetoni ha una energia di legame π maggiore di quella del legame σ, in contrato con la relazione π-σ nel legame C=C. Questo suggerisce che le reazioni di addizione ai gruppi carbonile dovrebbero essere termodinamicamente sfavoriti, come nel caso dell'addizione di acqua. Tutto questo è riassunto nel seguente schema (i valori di ΔH° sono per la reazione di addizione).
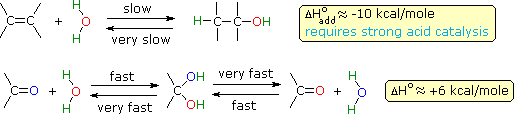 Sebbene l'addizione di acqua ad un alchene sia esotermica e dia un prodotto stabile (un alcole), la reazione non catalizzata è estremamente lenta a causa dell'elevata energia di attivazione. La reazione inversa (disidratazione di un alcole) è ancora più lenta, e a causa della barriera cinetica, entrambe le reazioni sono pratiche solo in presenza di un forte acido. Il meccanismo microscopicamente reversibile per entrambe le reazioni è stato discusso in precedenza, parlando degli alcoli.
Al contrario, sia l'addizione endotermica di acqua ad una funzione carbonilica, sia l'eliminazione esotermica di acqua dal risultante diolo geminale sono veloci. La polarità propria del gruppo carbonile, assieme alla sua aumentata basicità (comparata agli alcheni), abbassa l'energia dello stato di transizione per entrambe le reazioni, con un conseguente aumento nella velocità. Gli acidi e le basi catalizzano sia l'addizione che l'eliminazione di acqua. La prova che la rapida e reversibile addizione di acqua ai composti carbonilici avviene è fornita tramite esperimenti che usano acqua marca isotopicamente. Se un reagente carbonilico composto di 16O (colorato in blu sopra) viene trattato con acqua che incorpora l'isotopo 18O (colorato in rosso sopra), avviene un rapido scambio degli isotopi dell'ossigeno. Questo può essere spiegato solo tramite il meccanismo di addizione-eliminazione mostrato qui.
Sebbene l'addizione di acqua ad un alchene sia esotermica e dia un prodotto stabile (un alcole), la reazione non catalizzata è estremamente lenta a causa dell'elevata energia di attivazione. La reazione inversa (disidratazione di un alcole) è ancora più lenta, e a causa della barriera cinetica, entrambe le reazioni sono pratiche solo in presenza di un forte acido. Il meccanismo microscopicamente reversibile per entrambe le reazioni è stato discusso in precedenza, parlando degli alcoli.
Al contrario, sia l'addizione endotermica di acqua ad una funzione carbonilica, sia l'eliminazione esotermica di acqua dal risultante diolo geminale sono veloci. La polarità propria del gruppo carbonile, assieme alla sua aumentata basicità (comparata agli alcheni), abbassa l'energia dello stato di transizione per entrambe le reazioni, con un conseguente aumento nella velocità. Gli acidi e le basi catalizzano sia l'addizione che l'eliminazione di acqua. La prova che la rapida e reversibile addizione di acqua ai composti carbonilici avviene è fornita tramite esperimenti che usano acqua marca isotopicamente. Se un reagente carbonilico composto di 16O (colorato in blu sopra) viene trattato con acqua che incorpora l'isotopo 18O (colorato in rosso sopra), avviene un rapido scambio degli isotopi dell'ossigeno. Questo può essere spiegato solo tramite il meccanismo di addizione-eliminazione mostrato qui.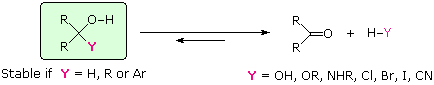 Se il sostituente Y non è un idrogeno, un gruppo alchile o un gruppo arile, c'è una buona possibilità che il composto sarà instabile (non isolabile), e decomporrà nella maniera mostrata. La maggior parte degli idrati e degli emiacetali (Y = OH & OR), per esempio, sono noti decomporre spontaneamente ai corrispondenti composti carbonilici. Gli aminoli (Y = NHR) sono intermedi nella formazione di un'immina, e anche essi ritornano ai loro precursori carbonilici se le condizioni di disidratazione non sono impiegate. Allo stesso modo, gli α-alogenoalcoli (Y = Cl, Br & I) non possono essere isolati, dato che essi si decompongono immediatamente con perdita di HY. In tutti e tre i casi l'addizione di H–Y ai gruppi carbonilici è chiaramente reversibile.
Se il sostituente Y è un idrogeno, un gruppo alchile o un gruppo arile, l'alcole risultante è un composto stabile e non si decompone con perdita di idrogeno o idrocarburi, anche per riscaldamente. Ne consegue quindi che se reagenti nucleofili del tipo H:–, R:– o Ar:– so addizionano ad aldeidi o chetoni, gli alcoli prodotti da tali addizioni si formeranno irreversibilmente. Anioni liberi di questi tipo dovrebbero essere basi e nucleofili estremamente forti, ma la loro straordinaria reattività li renderebbe difficili da preparare ed usare. Fortunatamente, i derivati metallici di questi alchili, arili ed idruri sono disponibili, e permettono la loro addizione ai composti carbonilici.
A. Riduzione tramite idruri complessi di metalli
L'addizione di un anione idruro ad un'aldeide o chetone produrrebbe un anione alcossido, che per protonazione dovrebbe portare all'alcole corrispondente. Le aldeidi darebbero alcoli primari (come mostrato) e i chetoni darebbero alcoli secondari.
RCH=O + H:–
Se il sostituente Y non è un idrogeno, un gruppo alchile o un gruppo arile, c'è una buona possibilità che il composto sarà instabile (non isolabile), e decomporrà nella maniera mostrata. La maggior parte degli idrati e degli emiacetali (Y = OH & OR), per esempio, sono noti decomporre spontaneamente ai corrispondenti composti carbonilici. Gli aminoli (Y = NHR) sono intermedi nella formazione di un'immina, e anche essi ritornano ai loro precursori carbonilici se le condizioni di disidratazione non sono impiegate. Allo stesso modo, gli α-alogenoalcoli (Y = Cl, Br & I) non possono essere isolati, dato che essi si decompongono immediatamente con perdita di HY. In tutti e tre i casi l'addizione di H–Y ai gruppi carbonilici è chiaramente reversibile.
Se il sostituente Y è un idrogeno, un gruppo alchile o un gruppo arile, l'alcole risultante è un composto stabile e non si decompone con perdita di idrogeno o idrocarburi, anche per riscaldamente. Ne consegue quindi che se reagenti nucleofili del tipo H:–, R:– o Ar:– so addizionano ad aldeidi o chetoni, gli alcoli prodotti da tali addizioni si formeranno irreversibilmente. Anioni liberi di questi tipo dovrebbero essere basi e nucleofili estremamente forti, ma la loro straordinaria reattività li renderebbe difficili da preparare ed usare. Fortunatamente, i derivati metallici di questi alchili, arili ed idruri sono disponibili, e permettono la loro addizione ai composti carbonilici.
A. Riduzione tramite idruri complessi di metalli
L'addizione di un anione idruro ad un'aldeide o chetone produrrebbe un anione alcossido, che per protonazione dovrebbe portare all'alcole corrispondente. Le aldeidi darebbero alcoli primari (come mostrato) e i chetoni darebbero alcoli secondari.
RCH=O + H:–  RCH2O– + H3O+
RCH2O– + H3O+ 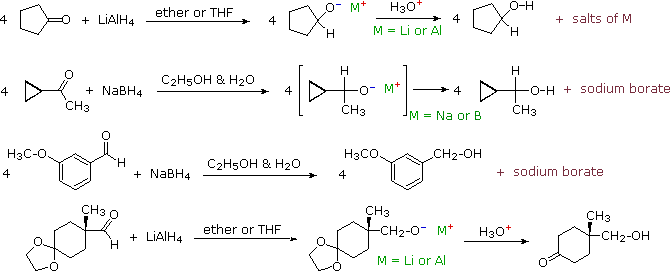 La riduzione di chetoni α,β-insaturi tramite reagenti idruri metallici a volte porta ad un alcole saturo, specialmente con sodio boroidruro. Questo prodotto è formato tramite un'addizione coniugata iniziale dell'idruro all'atomo di carbonio β, seguita da chetonizzazione dell'enolo prodotto e riduzione del risultante chetone saturo (equazione 1 sotto). Se l'alcole saturo è il prodotto desiderato, può essere necessaria l'idrogenazione catalitica prima (o dopo) la riduzione con idruro. Per evitare la riduzione del doppio legame, alla reazione viene aggiunto del cloruro di cerio(III) e la reazione è normalmente condotta al di sotto degli 0 °C, come mostrato nell'equazione 2.
1) RCH=CHCOR' + NaBH4 (alcole acquoso)
La riduzione di chetoni α,β-insaturi tramite reagenti idruri metallici a volte porta ad un alcole saturo, specialmente con sodio boroidruro. Questo prodotto è formato tramite un'addizione coniugata iniziale dell'idruro all'atomo di carbonio β, seguita da chetonizzazione dell'enolo prodotto e riduzione del risultante chetone saturo (equazione 1 sotto). Se l'alcole saturo è il prodotto desiderato, può essere necessaria l'idrogenazione catalitica prima (o dopo) la riduzione con idruro. Per evitare la riduzione del doppio legame, alla reazione viene aggiunto del cloruro di cerio(III) e la reazione è normalmente condotta al di sotto degli 0 °C, come mostrato nell'equazione 2.
1) RCH=CHCOR' + NaBH4 (alcole acquoso) 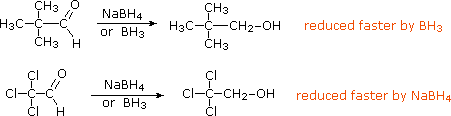 B. Addizione di reagenti organometallici
I due composti di questo tipo più comunemente usati sono gli alchil litio e i reagenti di Grignard. Essi vengono preparati da alogenuri alchilici ed arilici. Questi reagenti sono potenti nucleofili e basi davvero forti (le pKa degli idrocarburi saturi variano da 42 a 50), così essi si legano agli atomi di carbonio carbonilici, dando sali alcossido di litio o magnesio. A causa della loro tensione d'anello, gli epossidi subiscono molte reazioni tipo carboniliche, come notato precedentemente. Le reazioni di questo tipo sono tra i più importanti metodi sintetici disponibili al chimico, poiché essi permettono che semplici composti di partenza vengano uniti a formare strutture più complesse. Alcuni esempi sono mostrati nel seguente schema.
B. Addizione di reagenti organometallici
I due composti di questo tipo più comunemente usati sono gli alchil litio e i reagenti di Grignard. Essi vengono preparati da alogenuri alchilici ed arilici. Questi reagenti sono potenti nucleofili e basi davvero forti (le pKa degli idrocarburi saturi variano da 42 a 50), così essi si legano agli atomi di carbonio carbonilici, dando sali alcossido di litio o magnesio. A causa della loro tensione d'anello, gli epossidi subiscono molte reazioni tipo carboniliche, come notato precedentemente. Le reazioni di questo tipo sono tra i più importanti metodi sintetici disponibili al chimico, poiché essi permettono che semplici composti di partenza vengano uniti a formare strutture più complesse. Alcuni esempi sono mostrati nel seguente schema.
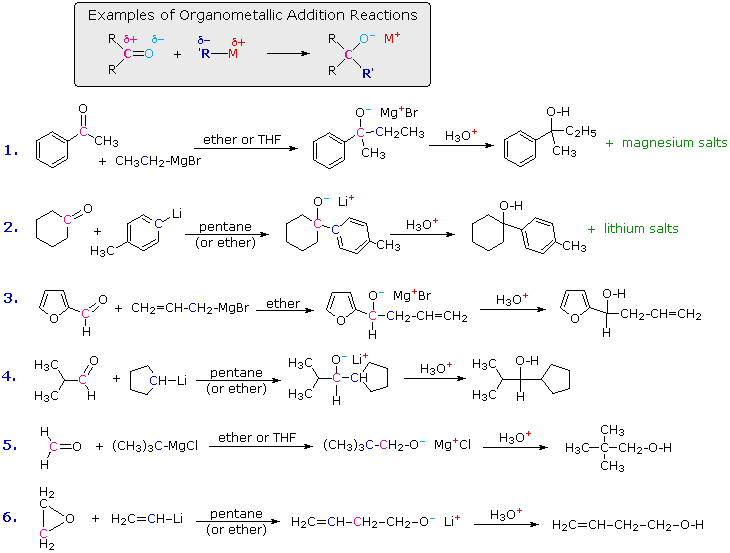 Un modello comune, mostrato nel riquadro ombreggiato in alto, è osservato in tutte queste reazioni. Il reagente organometallico è una fonte di un gruppo nucleofilo alchile o arile (colorato in blu), che si lega al carbonio elettrofilo del gruppo carbonile (colorato in magenta). Il prodotto di questa addizione è un sale alcossido metallico, e il prodotto alcole è generato tramite idrolisi acida debole del sale. I primi due esempi mostrano che anche i sali solubili in acqua di magnesio o litio vengono formati nell'idrolisi, ma questi sono raramente elencati tra i prodotti, come nelle ultime quattro reazioni. I chetoni reagiscono con i reagenti organometallici a dare alcoli terziari; la maggior parte delle aldeidi reagisce a dare alcoli secondari; e la formaldeide e l'ossido di etilene reagiscono formando alcoli primari (esempi #5 & 6). Quando un centro chirale è formato da reagenti achirali (esempi #1, 3 & 4) il prodotto è sempre una miscela racemica di enantiomeri.
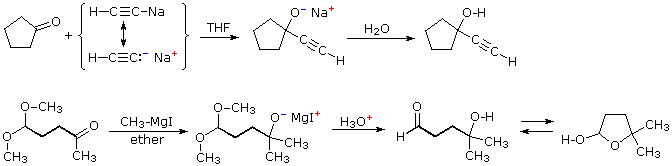
Due esempi addizionali dell'addizione di reagenti organometallici ai composti carbonilici sono informativi. Il primo dimostra che i derivati di metalli attivi della funzione terminale dell'alchino funzionano nello stesso modo di un alchil litio e dei reagenti di Grignard. Il secondo esempio illustra ancora l'uso dei gruppi protettivi acetalici in reazioni con potenti nucleofili. In seguito all'idrolisi acido-catalizzata dell'acetale, la risultante 4-idrossialdeide è in equilibrio con il suo emiacetale ciclico.
Un modello comune, mostrato nel riquadro ombreggiato in alto, è osservato in tutte queste reazioni. Il reagente organometallico è una fonte di un gruppo nucleofilo alchile o arile (colorato in blu), che si lega al carbonio elettrofilo del gruppo carbonile (colorato in magenta). Il prodotto di questa addizione è un sale alcossido metallico, e il prodotto alcole è generato tramite idrolisi acida debole del sale. I primi due esempi mostrano che anche i sali solubili in acqua di magnesio o litio vengono formati nell'idrolisi, ma questi sono raramente elencati tra i prodotti, come nelle ultime quattro reazioni. I chetoni reagiscono con i reagenti organometallici a dare alcoli terziari; la maggior parte delle aldeidi reagisce a dare alcoli secondari; e la formaldeide e l'ossido di etilene reagiscono formando alcoli primari (esempi #5 & 6). Quando un centro chirale è formato da reagenti achirali (esempi #1, 3 & 4) il prodotto è sempre una miscela racemica di enantiomeri.
Due esempi addizionali dell'addizione di reagenti organometallici ai composti carbonilici sono informativi. Il primo dimostra che i derivati di metalli attivi della funzione terminale dell'alchino funzionano nello stesso modo di un alchil litio e dei reagenti di Grignard. Il secondo esempio illustra ancora l'uso dei gruppi protettivi acetalici in reazioni con potenti nucleofili. In seguito all'idrolisi acido-catalizzata dell'acetale, la risultante 4-idrossialdeide è in equilibrio con il suo emiacetale ciclico.
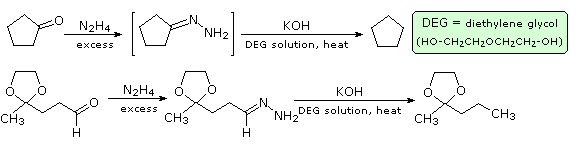 Il meccanismo di questa utile trasformazione comporta la tautomerizzazione dell'idrazione inizialmente formatosi in un isomero azo, che viene mostrato sotto nello schema meccanicistico. Le condizioni fortemente basiche usate in questa reazione precludono la tua applicazione a composti sensibili alle basi.
Il meccanismo di questa utile trasformazione comporta la tautomerizzazione dell'idrazione inizialmente formatosi in un isomero azo, che viene mostrato sotto nello schema meccanicistico. Le condizioni fortemente basiche usate in questa reazione precludono la tua applicazione a composti sensibili alle basi.
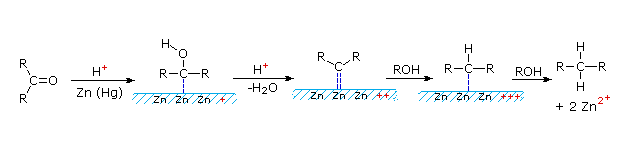
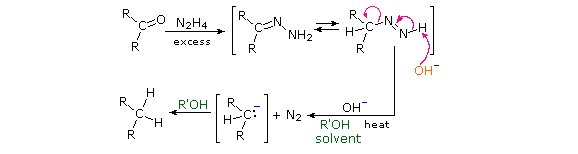 2. Riduzione di Clemmensen
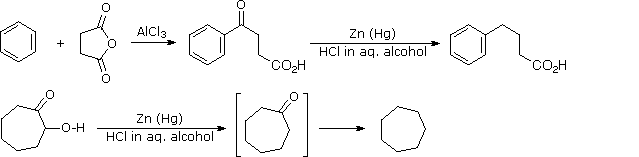
Questa riduzione alternativa comporta il riscaldamento di un composto carbonilico in presenza di zinco amalgamato, finemente suddiviso in un solvente idrossilico (spesso una miscela acquosa) contenente un acido minerale come HCl. Il mercurio in lega con lo zinco non partecipa alla reazione, serve solo a fornire un superficie metallica attiva pulita. Il primo esempio sotto mostra un'applicazione comune di questa riduzione, la conversione di un prodotto di acilazione di Friedel-Crafts in una catena laterale alchilica. Il secondo esempio illustra la labilità dei sostituenti funzionali in α ad un gruppo carbonile. I sostituenti come l'idrossile, l'alcossile & gli alogeni vengono ridotti per primi, la risultante aldeide o il chetone non sostituito vengono quindi ridotti all'idrocarburo corrispondente.
2. Riduzione di Clemmensen
Questa riduzione alternativa comporta il riscaldamento di un composto carbonilico in presenza di zinco amalgamato, finemente suddiviso in un solvente idrossilico (spesso una miscela acquosa) contenente un acido minerale come HCl. Il mercurio in lega con lo zinco non partecipa alla reazione, serve solo a fornire un superficie metallica attiva pulita. Il primo esempio sotto mostra un'applicazione comune di questa riduzione, la conversione di un prodotto di acilazione di Friedel-Crafts in una catena laterale alchilica. Il secondo esempio illustra la labilità dei sostituenti funzionali in α ad un gruppo carbonile. I sostituenti come l'idrossile, l'alcossile & gli alogeni vengono ridotti per primi, la risultante aldeide o il chetone non sostituito vengono quindi ridotti all'idrocarburo corrispondente.
 Un meccanismo possibile per la riduzione di Clemmensen è mostrato qui sotto.
Un meccanismo possibile per la riduzione di Clemmensen è mostrato qui sotto.
 3. Idrogenolisi dei tioacetali
Diversamente dalle precedenti due procedure, questo metodo di deossigenazione del carbonile richiede due passaggi separati. Deve, comunque, essere evitato il trattamento con acidi o basi forti.
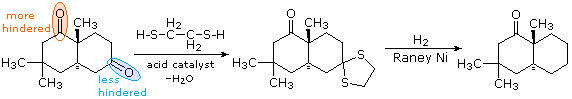
Il primo passaggio è la conversione dell'aldeide o del chetone in un tioacetale, come descritto in precedenza. Questi dewrivati possono essere isolati e purificati prima di continuare la riduzione. Il secondo passaggio comporta il riflussare una soluzione in acetone del tioacetale sopra un catalizzatore di nichel reattivo, chiamato Nichel Raney. Tutti i legami carbonio-zolfo subiscono idrogenolisi (i legami C–S vengono rotti tramite addizione di idrogeno). Nel seguente esempio, l'1,2-etanditiolo viene usato per preparare il tioacetale intermedio, a causa dell'elevata resa che questo reagente di solito fornisce. Il composto biciclico mostrato qui ha due gruppi carbonile, uno dei quali è stericamente ingombrato (cerchiato in arancione). Di conseguenza, un mono-tioacetale è facilmente preparato dal chetone meno ingombrato, e questi viene ridotto senza cambiare la funzione carbonilica rimanente.
3. Idrogenolisi dei tioacetali
Diversamente dalle precedenti due procedure, questo metodo di deossigenazione del carbonile richiede due passaggi separati. Deve, comunque, essere evitato il trattamento con acidi o basi forti.
Il primo passaggio è la conversione dell'aldeide o del chetone in un tioacetale, come descritto in precedenza. Questi dewrivati possono essere isolati e purificati prima di continuare la riduzione. Il secondo passaggio comporta il riflussare una soluzione in acetone del tioacetale sopra un catalizzatore di nichel reattivo, chiamato Nichel Raney. Tutti i legami carbonio-zolfo subiscono idrogenolisi (i legami C–S vengono rotti tramite addizione di idrogeno). Nel seguente esempio, l'1,2-etanditiolo viene usato per preparare il tioacetale intermedio, a causa dell'elevata resa che questo reagente di solito fornisce. Il composto biciclico mostrato qui ha due gruppi carbonile, uno dei quali è stericamente ingombrato (cerchiato in arancione). Di conseguenza, un mono-tioacetale è facilmente preparato dal chetone meno ingombrato, e questi viene ridotto senza cambiare la funzione carbonilica rimanente.
 B. Ossidazione
L'atomo di carbonio di un gruppo carbonilico ha uno stato di ossidazione relativamente alto. Questo è riflesso nel fatto che la maggior parte delle reazioni descritte finora o non causano un cambiamento nello stato di ossidazione (ad es. formazione di acetale ed immina) o effettuano una riduzione (ad es. addizioni organometalliche e deossigenazioni). La reazione di ossidazione più comune e caratteristica è la conversione di aldeidi ad acidi carbossilici. Nell'equazione stenografica mostrata qui il simbolo [O] si riferisce a condizioni di ossidazione non specificate che effettuano il cambiamento desiderato. Alcuni diversi metodi di effettuare questa trasformazione saranno qui descritti.
RCH=O + [O]
B. Ossidazione
L'atomo di carbonio di un gruppo carbonilico ha uno stato di ossidazione relativamente alto. Questo è riflesso nel fatto che la maggior parte delle reazioni descritte finora o non causano un cambiamento nello stato di ossidazione (ad es. formazione di acetale ed immina) o effettuano una riduzione (ad es. addizioni organometalliche e deossigenazioni). La reazione di ossidazione più comune e caratteristica è la conversione di aldeidi ad acidi carbossilici. Nell'equazione stenografica mostrata qui il simbolo [O] si riferisce a condizioni di ossidazione non specificate che effettuano il cambiamento desiderato. Alcuni diversi metodi di effettuare questa trasformazione saranno qui descritti.
RCH=O + [O] 
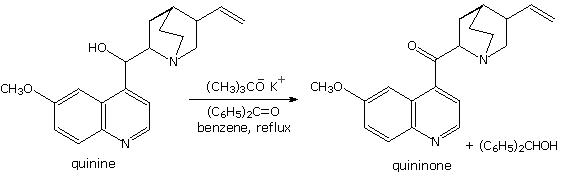
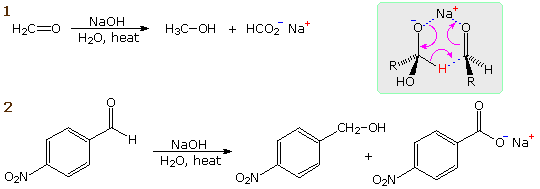
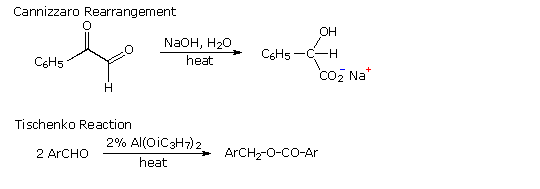
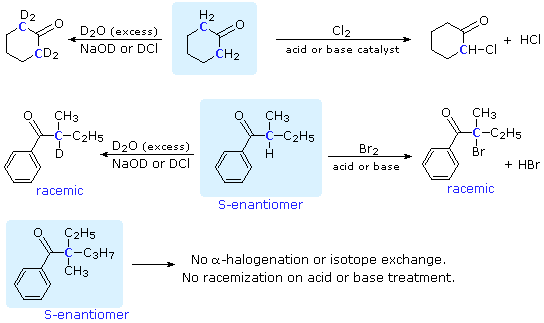
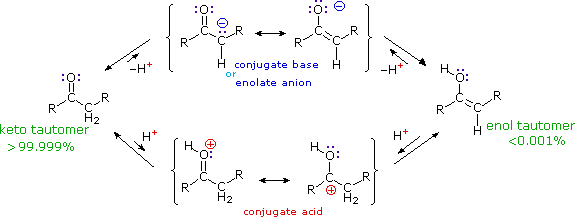
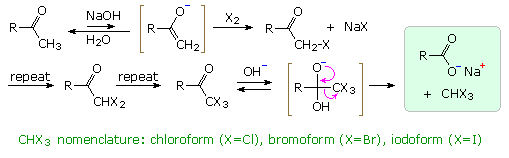
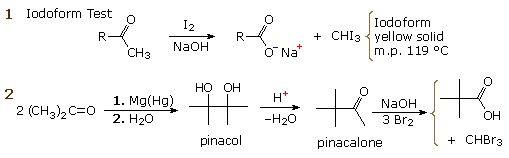
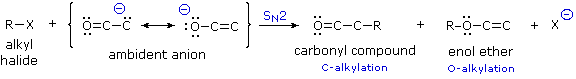
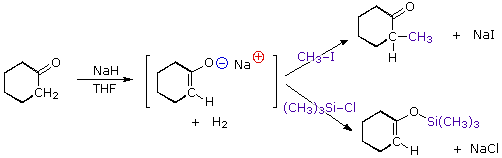
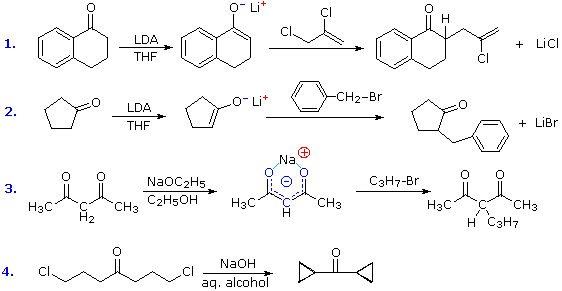
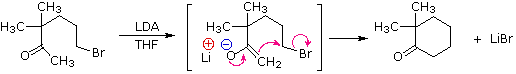
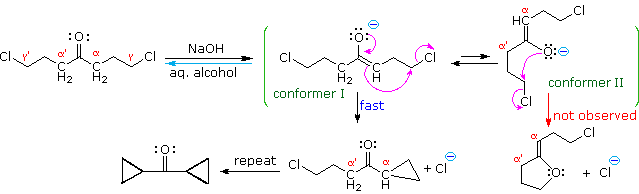 La catena a cinque atomi del diclorochetone può adottare molte conformazioni, due delle quali sono approssimante nel precedente schema. Sebbene il conformero II dell'anione enolato possa generare uno stabile anello a cinque membri tramite una reazione intramolecolare SN2, assumendo l'orientazione corretta degli atomi di carbonio α e γ', la concentrazione di questa struttura idealmente a spirale sarà molto bassa. In questo caso l'O-alchilazione dell'anione enolato, piuttosto che la C-alchilazione, è preferita per ragioni stereoelettroniche (regole di Baldwin). D'altra parte, le conformazioni in cui i carboni α e γ sono allineati correttamente per la formazione dell'anello a tre membri sono molto più numerose, ed il risultato è che più velocemente viene formata la base più essa subisce una ciclizzazione rapida ed irreversibile.
La chiusura d'anello a quattro, cinque, sei e sette membri sono possibili anch'esse tramtie alchilazione intramolecolare dell'enolato, come illustrato tramite il seguente esempio. In generale, gli anelli a cinque e sei membri sono termodinamicamente più stabili, mentre la formazione di anelli a tre membri è favorita cineticamente.
La catena a cinque atomi del diclorochetone può adottare molte conformazioni, due delle quali sono approssimante nel precedente schema. Sebbene il conformero II dell'anione enolato possa generare uno stabile anello a cinque membri tramite una reazione intramolecolare SN2, assumendo l'orientazione corretta degli atomi di carbonio α e γ', la concentrazione di questa struttura idealmente a spirale sarà molto bassa. In questo caso l'O-alchilazione dell'anione enolato, piuttosto che la C-alchilazione, è preferita per ragioni stereoelettroniche (regole di Baldwin). D'altra parte, le conformazioni in cui i carboni α e γ sono allineati correttamente per la formazione dell'anello a tre membri sono molto più numerose, ed il risultato è che più velocemente viene formata la base più essa subisce una ciclizzazione rapida ed irreversibile.
La chiusura d'anello a quattro, cinque, sei e sette membri sono possibili anch'esse tramtie alchilazione intramolecolare dell'enolato, come illustrato tramite il seguente esempio. In generale, gli anelli a cinque e sei membri sono termodinamicamente più stabili, mentre la formazione di anelli a tre membri è favorita cineticamente.