quimico
2011-12-28 08:55
1. Nomenclatura e struttura
Nel sistema di nomenclatura IUPAC, i gruppi funzionali sono normalmente designati in uno di due modi. La presenza della funzionalità può essere indicata tramite un suffisso caratteristico ed un numero di locazione. Questo è comune per i doppi e tripli legami carbonio-carbonio che hanno i rispettivi suffissi -ene ed -ino. Gli alogeni, dall'altro lato, non hanno un suffisso e sono chiamati come sostituenti, per esempio: (CH3)2C=CHCHClCH3 è il 4-cloro-2-metil-2-pentene.
Le amine sono derivati dell'ammoniaca in cui uno o più degli idrogeni sono stati sostituiti da un gruppo alchile o arile. La nomenclatura delle amine è complicata dal datto che esistono molti diversi sistemi di nomenclatura, e non c'è una chiara preferenza per uno rispetto agli altri. Quindi, i termini primario (1º), secondario (2º) & terziario (3º) sono usati per classificare le amine in una maniera completamente diversa da quella usata per gli alcoli o gli alogenuri alchilici. Quando questi termini sono applicati alle amine si riferiscono al numero di sostituenti alchilici (o arilici) legati all'atomo di azoto, mentre negli altri casi si riferiscono alla natura del gruppo alchile.
I quattro composti mostrati nella fila superiore del seguente schema sono tutti isomeri di formula C4H11N. I primi due sono classificati come amine primarie, dato che solo un gruppo alchile è legato all'azoto; comunque, il gruppo alchile è primario nel primo esempio e terziario nel secondo. I composti terzo e quarto nella fila sono amine secondaria e terziaria rispettivamente. Un azoto legato a quattro gruppi alchile sarà necessariamente caricato positivamente, ed è chiamato un catione ammonio quaternario. Per esempio, il (CH3)4N+Br– è il tetrametilammonio bromuro.
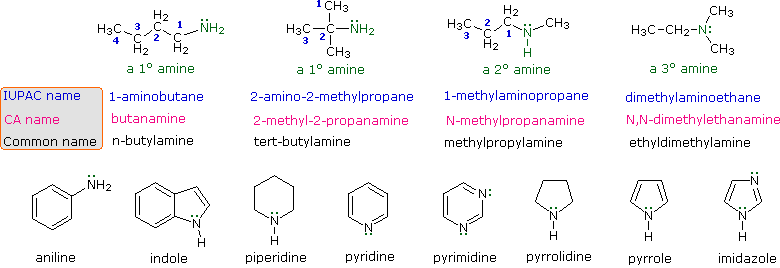
I nomi IUPAC sono elencati per primi e colorati in blu. Questo sistema nomina le funzioni aminiche come sostituenti sul più grande gruppo alchile. Il semplice sostituente -NH2 trovato nelle amine primario è chiamato gruppo amino. Per le amine secondarie e terziarie un prefisso del composto (ad esempio dimetilamino nel quarto esempio) include i nomi di tutti ma la radice del gruppo alchile.
Il servizio Chemical Abstract ha adottato un sistema di nomenclatura in cui il suffisso -amina è attaccato alla radice del nome dell'alchile. Per le amine primarie come la butanamina (primo esempio) questa è analoga alla nomenclatura IUPAC degli alcoli (suffisso -olo). I sostituenti addizionali all'azoto nelle amine secondarie e terziarie sono designati tramite il prefisso N- prima del nome del gruppo. Questi nomi CA sono colorati in magenta nello schema.
Infine, un sistema comune per amine semplici nomina ogni sostituente alchile sull'azoto in ordine alfabetico, seguito dal suffisso -amina. Questi sono i nomi riportati nell'ultima fila (colorati in nero).
Molte amine aromatiche ed eterocicliche sono note tramite nomi comuni unici, le cui origini sono spesso ignote ai chimici che le usano frequentemente. Dato che questi nomi non sono basati su un sistema razionale, è necessario memorizzarli. Come abbiamo già visto, esiste una nomenclatura sistematica per i composti eterociclici.
Composti azotati naturali
In natura abbondando i composti azotati, molti dei quali si ritrovano in piante e ci si riferisce ad essi col termine alcaloidi. Le formule strutturali per alcuni alcaloidi rappresentativi e per altri composti naturali azotati sono mostrate sotto, e possiamo riconoscere molte delle caratteristiche strutturali base elencate sopra nelle loro formule. Quindi, la serotonina e la tiamina sono amine primarie, la coniina è un'amina secondaria, l'atropina, la morfina e la chinina sono amine terziarie, e la muscarina è un sale di ammonio quaternario.
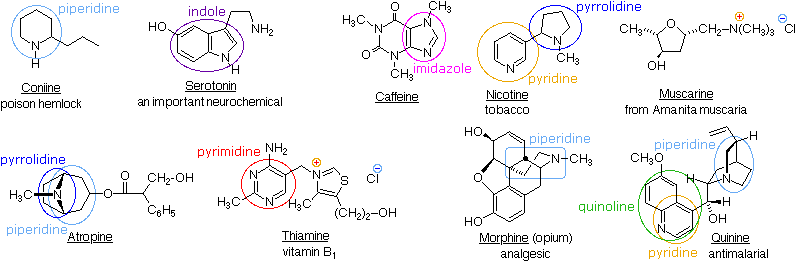
Gli atomi di azoto che sono parte degli anelli aromatici, come nella piridina, nel pirrolo, nell'imidazolo, hanno configurazioni planari (ibridazione sp2), e non sono centri stereogenici. Anche gli atomi di azoto legati a gruppi carbonilici, come nella caffeina, tendono ad essere planari. Invece, l'atropina, la coniina, la morfina, la nicotina e la chinina hanno atomi di azoto piramidali stereogenici nelle loro formule strutturali (pensate al doppietto elettronico non legante come ad un quarto sostituente su un atomo di azoto ibridizzato sp3). Nella chinina questo azoto è limitato ad una configurazione dal sistema ciclico pontato. Gli altri azoti stereogenici sono liberi di assumere due configurazioni piramidali, ma queste sono in rapido equilibrio così che non possono essere facilmente isolati degli stereoisomeri distinti che riflettano questi siti.
Dovrebbe essere notato che i fattori strutturali possono servire a permettere la risoluzione di amine chirali piramidali. Due esempi di tali amine terziarie, comparate ad analoghi non risolvibili, sono mostrati nel primo schema, sotto. I due atomi di azoto nella base di Trögers sono gli unici centri stereogenici nella molecola. A causa della struttura pontata della molecola, gli atomi di azoto hanno la stessa configurazione e non possono subire inversione. La cloro aziridina può invertire, ma per fare ciò è richiesta una energia di attivazione elevata, comparata con le più grandi amine eterocicliche. È stata infatti risolta, e sono stati isolati enantiomeri puri. Un aumento nella tensione angolare nello stato di transizione planare ibridizzato sp2 è responsabile per la maggiore stabilità della configurazione piramidale. La rozza stima della tensione angolare è fatta usando un angolo C-N-C di 60° come valore arbitrario per l'eterociclo a tre membri.
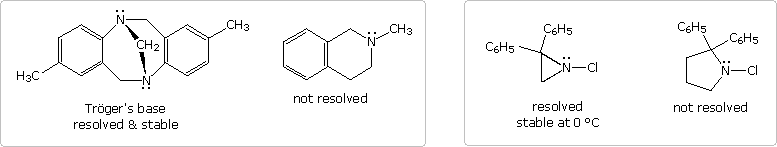

Ovviamente, i sali di ammonio quaternari, come quello nella muscarina, hanno una configurazione tetraedrica che è incapace di inversione. Con quattro differenti sostituenti, un tale azoto dovrebbe essere un centro stereogenico stabile.
I seguenti utenti ringraziano quimico per questo messaggio: jobba
 pKa 11.0
pKa 11.0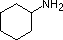 pKa 10.7
pKa 10.7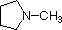 pKa 10.7
pKa 10.7 pKa 5.2
pKa 5.2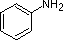 pKa 4.6
pKa 4.6 pKa 1.0
pKa 1.0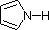 pKa 0.0
pKa 0.0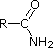 pKa -1.0
pKa -1.0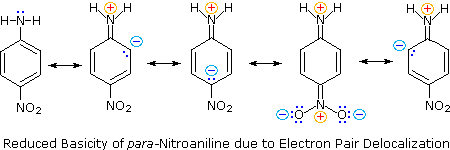
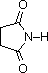 pKa 9.6
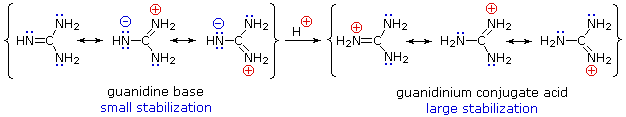
Gli acidi mostrati qui possono essere convertiti nelle loro basi coniugate con basi derivati da acidi più deboli (basi più forti). Tre esempi di tali reazioni sono mostrate sotto, con l'idrogeno acido colorato in rosso in ogni caso. Per la conversione completa nella base coniugata, come mostrato, è richiesta come reagente un base circa un milione di volte più forte.
C6H5SO2NH2 + KOH → C6H5SO2NH–K+ + H2O una base solfonammide
(CH3)3COH + NaH → (CH3)3CO–Na+ + H2 un base alcossido
(C2H5)2NH + C4H9Li → (C2H5)2N–Li+ + C4H10 una base ammide
4. Importanti basi reagenti
Il significato di tutte queste reazioni acido-base verso la chimica organica pratica giace nella necessità di basi organiche di forza mutevole, come i reagenti fatti su misura a seconda delle richieste di reazioni specifiche. La comune base sodio idrossido non è solubile in molti solventi organici, e non è quindi diffusamente usata come reagente nelle reazioni organiche. La maggiro parte delle basi usate come reagenti sono sali di alcossidi, amine o ammidi. Dato che gli alcoli sono acidi molto più forti delle amine, le loro basi coniugate sono più deboli delle basi ammidiche, e riempiono la distanza nella forza della base tra i sali delle amine e delle ammidi. Nell'elenco sotto, ancora le pKa si riferiscono all'acido coniugato della base disegnata sopra di esse.
Piridina
pKa 9.6
Gli acidi mostrati qui possono essere convertiti nelle loro basi coniugate con basi derivati da acidi più deboli (basi più forti). Tre esempi di tali reazioni sono mostrate sotto, con l'idrogeno acido colorato in rosso in ogni caso. Per la conversione completa nella base coniugata, come mostrato, è richiesta come reagente un base circa un milione di volte più forte.
C6H5SO2NH2 + KOH → C6H5SO2NH–K+ + H2O una base solfonammide
(CH3)3COH + NaH → (CH3)3CO–Na+ + H2 un base alcossido
(C2H5)2NH + C4H9Li → (C2H5)2N–Li+ + C4H10 una base ammide
4. Importanti basi reagenti
Il significato di tutte queste reazioni acido-base verso la chimica organica pratica giace nella necessità di basi organiche di forza mutevole, come i reagenti fatti su misura a seconda delle richieste di reazioni specifiche. La comune base sodio idrossido non è solubile in molti solventi organici, e non è quindi diffusamente usata come reagente nelle reazioni organiche. La maggiro parte delle basi usate come reagenti sono sali di alcossidi, amine o ammidi. Dato che gli alcoli sono acidi molto più forti delle amine, le loro basi coniugate sono più deboli delle basi ammidiche, e riempiono la distanza nella forza della base tra i sali delle amine e delle ammidi. Nell'elenco sotto, ancora le pKa si riferiscono all'acido coniugato della base disegnata sopra di esse.
Piridina 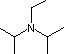 pKa 11.4
DBU
pKa 11.4
DBU 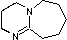 pKa 12
Base di Barton
pKa 12
Base di Barton 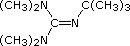 pKa 14
Potassio t-butossido (CH3)3CO–K+ pKa 19
Sodium HMDS [(CH3)3Si]2N–Na+ pKa 26
LDA [(CH3)2CH]2N–Li+ pKa 35.7
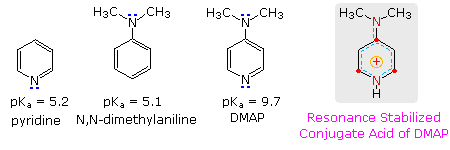
La piridina è comunemente usta come scavenger di acido in reazioni che producono co-prodotti acidi minerali. La sua basicità e nucleofilicità può essere modificata tramite ingombro sterico, come nel caso della 2,6-dimetilpiridina (2,6-lutidina) (pKa = 6.7), o tramite stabilizzazione per risonanza, come nel caso della 4-dimetilaminopiridina (pKa = 9.7). La base di Hünig è relativamente non nucleofila (a causa dell'ingombro sterico), e come il DBU è spesso usata come base nelle reazioni di eliminazione E2 condotte in solventi non polari. La base di Barton è una base forte, debolmente nucleofila, neutra che serve in casi dove la sostituzione elettrofila del DBU o di altre basi aminiche è un problema. Gli alcossidi sono basi più forti che sono spesso usate nei corrispondenti alcoli come solventi, o per maggiore reattività in DMSO. Infine, le due basi ammidiche sono usati diffusamente nella generazione di enolati da composti carbonilici ed altri carboni acidi deboli.
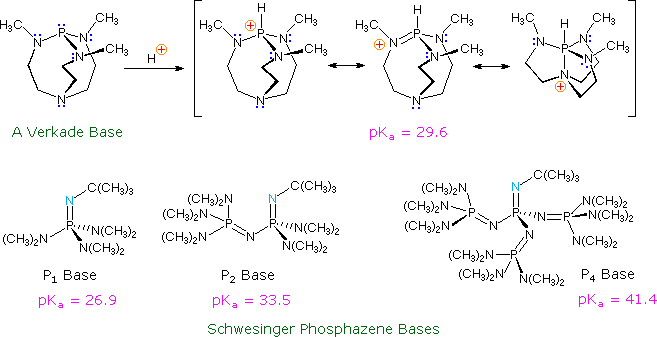
Superbasi non ioniche
Le più foti basi disponibili per i chimici organici sono i sali alcossidici dei metalli alcalini, come il KOBut, gli alchili dei metalli alcalini, come il nBuLi ed i sali ammidici dei metalli alcalini, come l'LDA. Queste potenti basi sono tutte potenziali nucleofili (alcune più delle altre) ed hanno legami parzialmente ionici col metallo. Recentemente, una nuova classe di basi neutre, non metalliche, debolmente nucleofile è stata preparata e studiata. Alcuni esempi sono mostrati nel seguente schema.
Il sito basico nella base di Verkade è l'atomo di fosforo, essendo l'acido coniugato stabilizzato tramite legame transannulare con l'azoto. La forza di queste basi può essere modificata dai sostituenti sugli azoti a fianco. Le base di Schwesinger, fosfazeniche aumentano la loro forza non appena altre unità fosfazeniche vengono aggiunte in coniugazione col sito basico (l'atomo di azoto blu chiaro).Tutte le pKa per queste basi sono misurate in acetonitrile.
pKa 14
Potassio t-butossido (CH3)3CO–K+ pKa 19
Sodium HMDS [(CH3)3Si]2N–Na+ pKa 26
LDA [(CH3)2CH]2N–Li+ pKa 35.7
La piridina è comunemente usta come scavenger di acido in reazioni che producono co-prodotti acidi minerali. La sua basicità e nucleofilicità può essere modificata tramite ingombro sterico, come nel caso della 2,6-dimetilpiridina (2,6-lutidina) (pKa = 6.7), o tramite stabilizzazione per risonanza, come nel caso della 4-dimetilaminopiridina (pKa = 9.7). La base di Hünig è relativamente non nucleofila (a causa dell'ingombro sterico), e come il DBU è spesso usata come base nelle reazioni di eliminazione E2 condotte in solventi non polari. La base di Barton è una base forte, debolmente nucleofila, neutra che serve in casi dove la sostituzione elettrofila del DBU o di altre basi aminiche è un problema. Gli alcossidi sono basi più forti che sono spesso usate nei corrispondenti alcoli come solventi, o per maggiore reattività in DMSO. Infine, le due basi ammidiche sono usati diffusamente nella generazione di enolati da composti carbonilici ed altri carboni acidi deboli.
Superbasi non ioniche
Le più foti basi disponibili per i chimici organici sono i sali alcossidici dei metalli alcalini, come il KOBut, gli alchili dei metalli alcalini, come il nBuLi ed i sali ammidici dei metalli alcalini, come l'LDA. Queste potenti basi sono tutte potenziali nucleofili (alcune più delle altre) ed hanno legami parzialmente ionici col metallo. Recentemente, una nuova classe di basi neutre, non metalliche, debolmente nucleofile è stata preparata e studiata. Alcuni esempi sono mostrati nel seguente schema.
Il sito basico nella base di Verkade è l'atomo di fosforo, essendo l'acido coniugato stabilizzato tramite legame transannulare con l'azoto. La forza di queste basi può essere modificata dai sostituenti sugli azoti a fianco. Le base di Schwesinger, fosfazeniche aumentano la loro forza non appena altre unità fosfazeniche vengono aggiunte in coniugazione col sito basico (l'atomo di azoto blu chiaro).Tutte le pKa per queste basi sono misurate in acetonitrile.
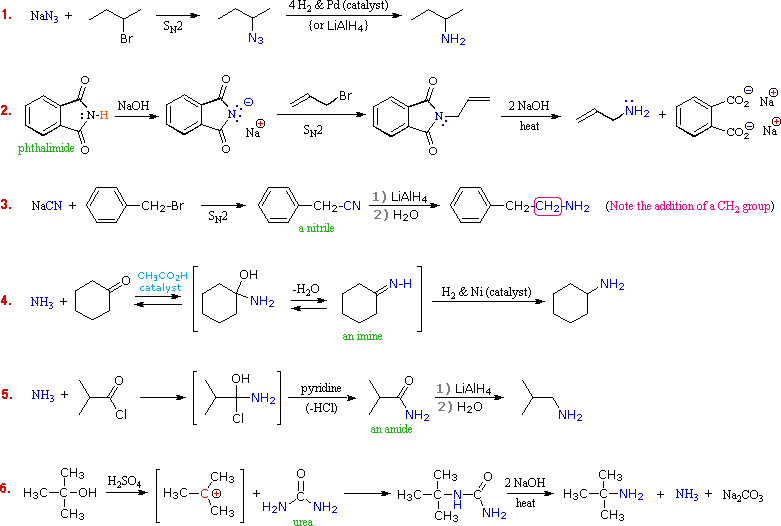
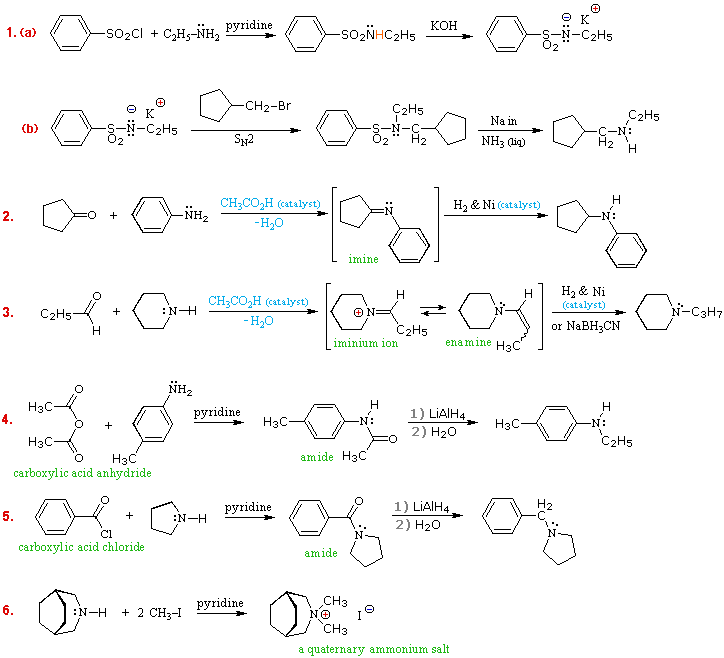
 R2N̈HE+
R2N̈HE+  RCH2NH2 + NH4+Br–
RCH2NH2 + NH4+Br–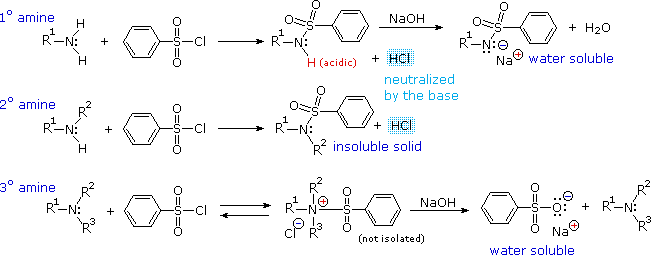
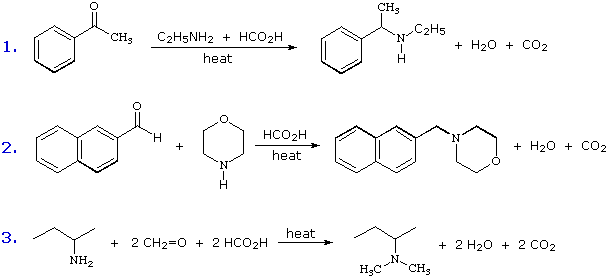
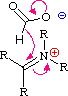
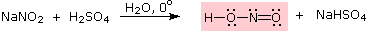
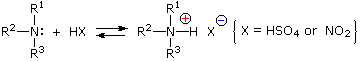

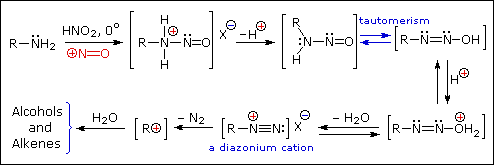
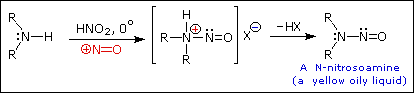
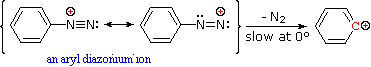
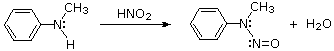
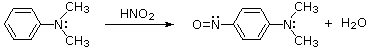
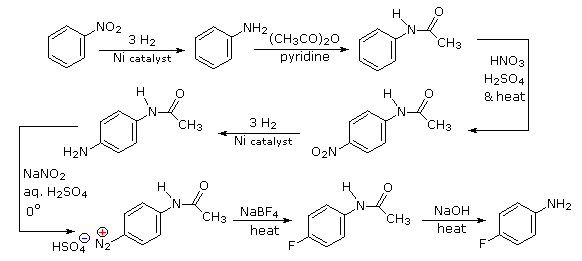
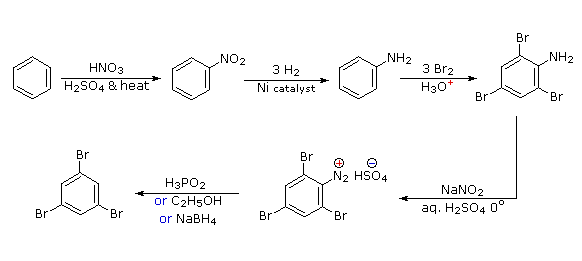
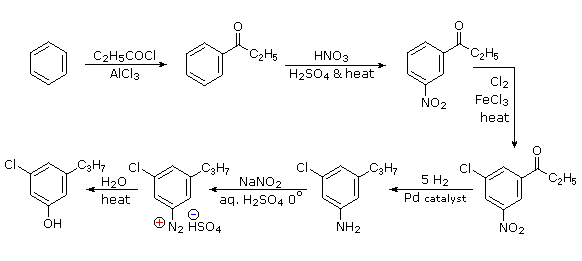
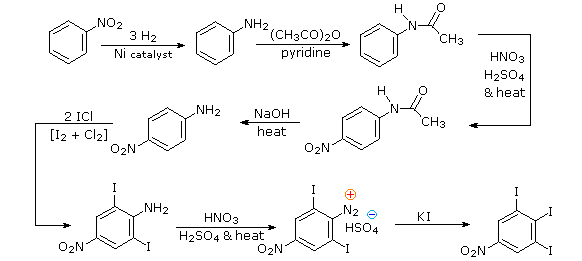
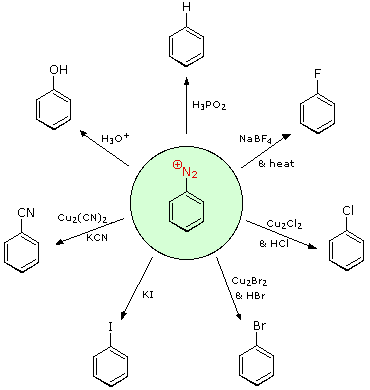 Queste reazioni di sostituzione degli aril diazonio espandono in modo significativo le tattiche disponibili per la sintesi di derivati del benzene polisostituiti. Considerate le seguenti opzioni:
(i) Il tipico precursore di un amina arilica è il corrispondente nitro composto. Un sostituente nitro disattiva un anello aromatico e direziona la sostituzione elettrofila verso le posizioni meta.
(ii) La riduzione di un gruppo nitro in un'amina può essere ottenuta in diversi modi. Il risultante sostituente amino attiva fortemente un anello aromatico e direziona la sostituzione elettrofila verso le posizioni orto & para.
(iii) Il carattere attivante di un sostituente amino può essere attenuato tramite formazione di un derivato ammidico (reversibile), o anche essere cambiato in disattivamente e meta orientante tramite formazione di un sale di ammonio quaternario (irreversibile).
(iv) La conversione di un'amina arilica in un ione diazonio intermedio le permette di essere sostituita da una varietà di gruppi differenti (incluso l'idrogeno), che a loro volta possono essere usati per reazioni successive.
Queste reazioni di sostituzione degli aril diazonio espandono in modo significativo le tattiche disponibili per la sintesi di derivati del benzene polisostituiti. Considerate le seguenti opzioni:
(i) Il tipico precursore di un amina arilica è il corrispondente nitro composto. Un sostituente nitro disattiva un anello aromatico e direziona la sostituzione elettrofila verso le posizioni meta.
(ii) La riduzione di un gruppo nitro in un'amina può essere ottenuta in diversi modi. Il risultante sostituente amino attiva fortemente un anello aromatico e direziona la sostituzione elettrofila verso le posizioni orto & para.
(iii) Il carattere attivante di un sostituente amino può essere attenuato tramite formazione di un derivato ammidico (reversibile), o anche essere cambiato in disattivamente e meta orientante tramite formazione di un sale di ammonio quaternario (irreversibile).
(iv) La conversione di un'amina arilica in un ione diazonio intermedio le permette di essere sostituita da una varietà di gruppi differenti (incluso l'idrogeno), che a loro volta possono essere usati per reazioni successive.
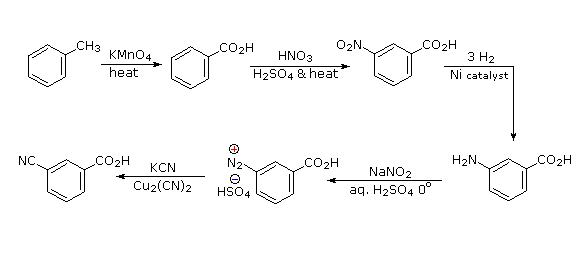
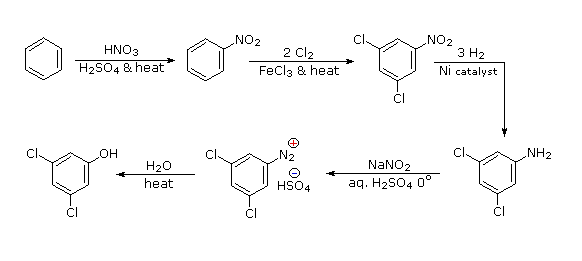
 Legame all'azoto
Una descrizione tramite risonanza degli ioni diazonio mostra che la carica positiva è delocalizzata sui due atomi di azoto. Non è possibile per i nucleofili legarsi all'azoto più interno, ma il legame (o il coupling) dei nucleofili negativi all'azoto terminale porta agli azo composti neutri. Come mostrato nella seguente equazione, questo coupling con l'azoto terminale dovrebbe essere relativamente veloce e reversibile. I prodotti azo possono esistere come stereoisomeri E/Z. In pratica è stato trovato che l'isomero E predomina all'equilibrio.
Legame all'azoto
Una descrizione tramite risonanza degli ioni diazonio mostra che la carica positiva è delocalizzata sui due atomi di azoto. Non è possibile per i nucleofili legarsi all'azoto più interno, ma il legame (o il coupling) dei nucleofili negativi all'azoto terminale porta agli azo composti neutri. Come mostrato nella seguente equazione, questo coupling con l'azoto terminale dovrebbe essere relativamente veloce e reversibile. I prodotti azo possono esistere come stereoisomeri E/Z. In pratica è stato trovato che l'isomero E predomina all'equilibrio.
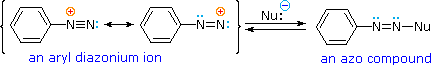 Nonostante questi prodotti azo siano intrappolati o stabilizzati in qualche modo, il ritorno a ione diazonio e la lenta sostituzione nucleofila al cabonio (con perdita irreversibile dell'azoto) saranno la direzione finale della reazione, come descritto nella sezione precedente. Per esempio, se il fenildiazonio bisolfato è aggiunto rapidamente ad una soluzione fredda di sodio idrossido una soluzione relativamente stabile di sodio fenildiazoato (la base coniugata dell'acido diazoico inizialmente formatosi) viene ottenuta. Abbassando il pH di questa soluzione si rigenera l'acido fenildiazoico (pKa ca. 7), che dissocia ritornando indietro a ione diazonio ed eventualmente subendo sostituzione, a generare fenolo.
C6H5N2+HSO4– + NaOH (soluzione fredda)
Nonostante questi prodotti azo siano intrappolati o stabilizzati in qualche modo, il ritorno a ione diazonio e la lenta sostituzione nucleofila al cabonio (con perdita irreversibile dell'azoto) saranno la direzione finale della reazione, come descritto nella sezione precedente. Per esempio, se il fenildiazonio bisolfato è aggiunto rapidamente ad una soluzione fredda di sodio idrossido una soluzione relativamente stabile di sodio fenildiazoato (la base coniugata dell'acido diazoico inizialmente formatosi) viene ottenuta. Abbassando il pH di questa soluzione si rigenera l'acido fenildiazoico (pKa ca. 7), che dissocia ritornando indietro a ione diazonio ed eventualmente subendo sostituzione, a generare fenolo.
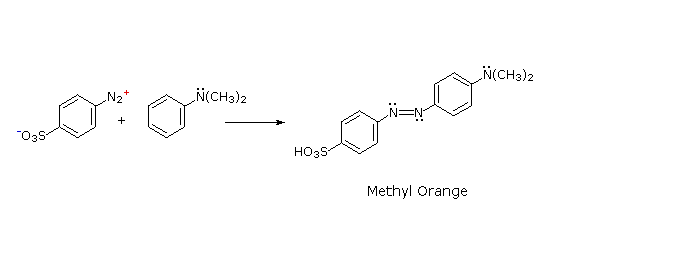
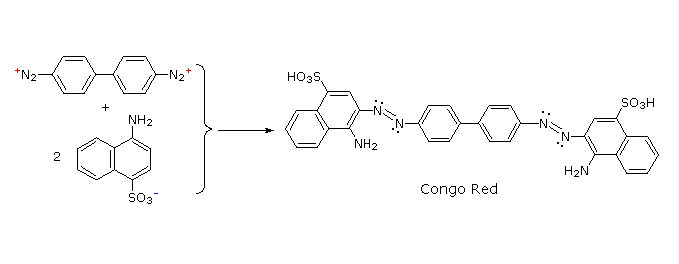
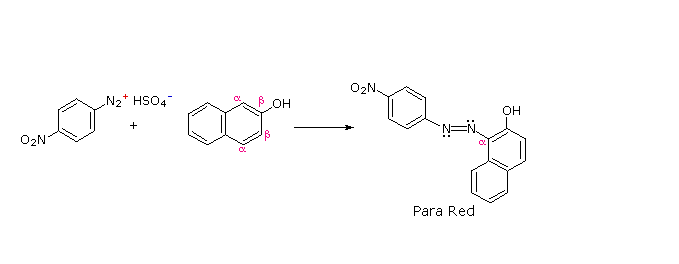
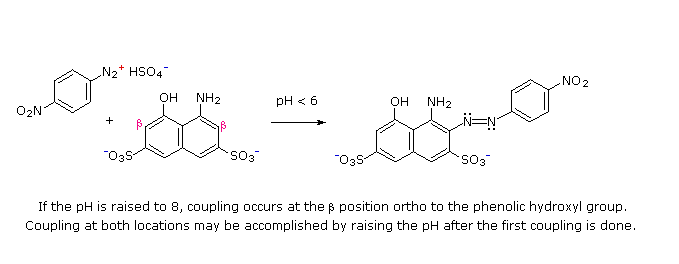
C6H5N2+HSO4– + NaOH (soluzione fredda) 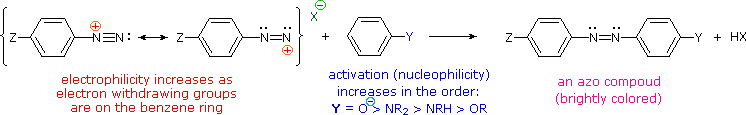 Alcuni esempi di reazioni di coupling per l'ottenimento di azo composti sono mostrate sotto. Poche semplici regole sono d'aiuto nel predirre il decorso di tali reazioni:
(i) A pH acido (< 6) un gruppo amino è un sostituente attivante più forte rispetto ad un gruppo idrossile (cioè un fenolo). A pH basico (> 7.5) le funzioni fenoliche sono attivanti più forti, a causa dell'aumentata concentrazione della base fenossido o fenolato.
(ii) Il coupling con un anello benzenico attivato avviene preferenzialmente in para rispetto al gruppo attivante se quella posizione è libera. In caso contrario avverrà un coupling in posizione orto.
(iii) Il naftalene normalmente subisce sostituzione elettrofila in posizione α più rapidamente rispetto ai siti β; comunque, il coupling in orto è preferito. Vedete lo schema per gli esempi della notazione α/β nei naftaleni.
Alcuni esempi di reazioni di coupling per l'ottenimento di azo composti sono mostrate sotto. Poche semplici regole sono d'aiuto nel predirre il decorso di tali reazioni:
(i) A pH acido (< 6) un gruppo amino è un sostituente attivante più forte rispetto ad un gruppo idrossile (cioè un fenolo). A pH basico (> 7.5) le funzioni fenoliche sono attivanti più forti, a causa dell'aumentata concentrazione della base fenossido o fenolato.
(ii) Il coupling con un anello benzenico attivato avviene preferenzialmente in para rispetto al gruppo attivante se quella posizione è libera. In caso contrario avverrà un coupling in posizione orto.
(iii) Il naftalene normalmente subisce sostituzione elettrofila in posizione α più rapidamente rispetto ai siti β; comunque, il coupling in orto è preferito. Vedete lo schema per gli esempi della notazione α/β nei naftaleni.
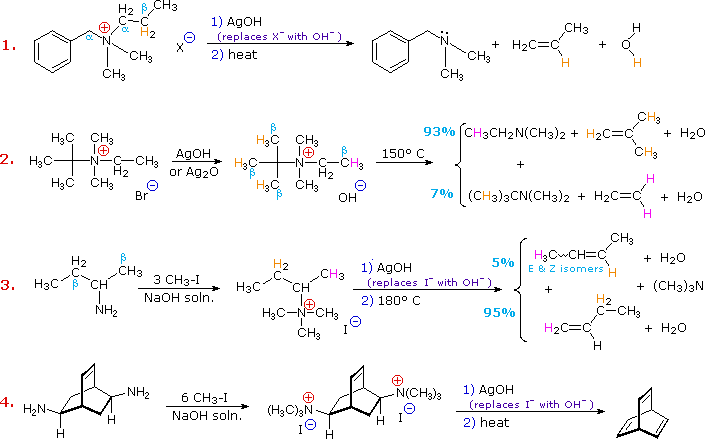
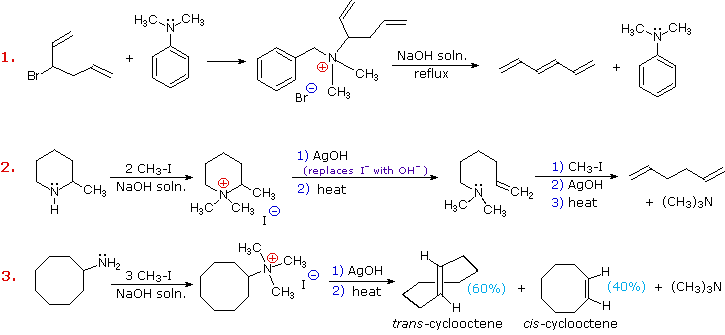
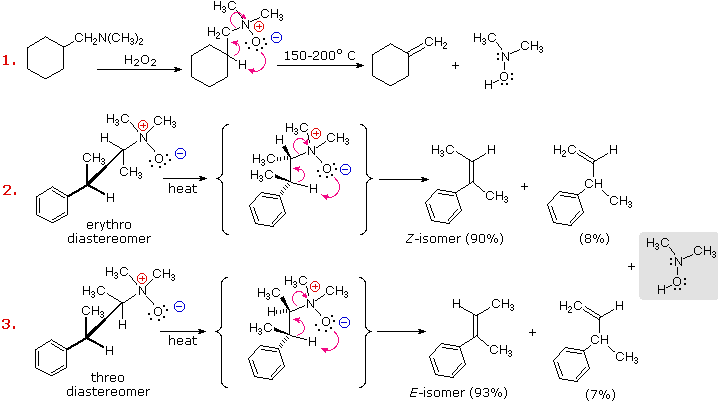 L'eliminazione di Cope di amine N-ossido diastereoisomeriche, come quelle mostrate negli esempi #2 & 3 sopra, forniscono la prova della relazione syn dei gruppi idrogeno β ed amina N-ossido. Questi esempi dimostrano anche una forte regioselettività che favorisce il doppio legame più stabile.
Radicali nitrossido
Le amine secondarie che non possiedono idrogeni α vengono ossidate dai perossidi (ZOOH) a radicali nitrossido di sorprendente stabilità. Nell'esempio mostrato nella parte alta del diagramma seguente andrebbe notato che la delocalizzazione per risonanza dell'elettrone non accoppiato contribuisce ad un legame polare N–O. Il composto con R = H, noto tramite l'acronimo TEMPO, è un solido rosso relativamente stabile. Sono stati preparati molti altri nitrossidi, tre dei quali sono disegnati nella parte bassa dello schema, a destra. Se uno o più idrogeni sono presenti su un carbonio adiacente, il nitrossido decompone in miscele che includono amine N-ossido e nitroni, come mostrato nella parte bassa dello schema, a sinistra. I nitrossidi sono ossidati dagli alogeni a cationi ossammonio instabili.
L'eliminazione di Cope di amine N-ossido diastereoisomeriche, come quelle mostrate negli esempi #2 & 3 sopra, forniscono la prova della relazione syn dei gruppi idrogeno β ed amina N-ossido. Questi esempi dimostrano anche una forte regioselettività che favorisce il doppio legame più stabile.
Radicali nitrossido
Le amine secondarie che non possiedono idrogeni α vengono ossidate dai perossidi (ZOOH) a radicali nitrossido di sorprendente stabilità. Nell'esempio mostrato nella parte alta del diagramma seguente andrebbe notato che la delocalizzazione per risonanza dell'elettrone non accoppiato contribuisce ad un legame polare N–O. Il composto con R = H, noto tramite l'acronimo TEMPO, è un solido rosso relativamente stabile. Sono stati preparati molti altri nitrossidi, tre dei quali sono disegnati nella parte bassa dello schema, a destra. Se uno o più idrogeni sono presenti su un carbonio adiacente, il nitrossido decompone in miscele che includono amine N-ossido e nitroni, come mostrato nella parte bassa dello schema, a sinistra. I nitrossidi sono ossidati dagli alogeni a cationi ossammonio instabili.
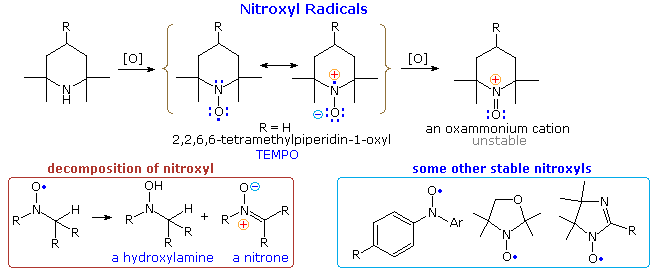 Lo spin dell'elettrone non accopiato del nitrosile può essere studiato tramite una tecnica chiamata EPR (Electron Paramagnetic Resonance) o ESR (Electron Spin Resonance). Esperimenti di questo tipo hanno dimostrato che gli spettri EPR sono sensibili ai sostituenti sul radicale così come al suo ambiente vicino. Questo ha portato ad una stategia di marcatura dello spin per l'indagine delle strutture conformazionali delle macromolecole come le proteine. percià, la tecnica site-directed spin labeling (SDSL) è emersa come tecnica preziosa per mappare gli elementi della struttura secondaria, al livello del fold del frammento, in un vasto range di proteine, incluse quelle non riconducibile alla caratterizzazione strutturale usando le classiche tecniche strutturali, quali l'NMR e la cristallografia ai raggi X.
Lo spin dell'elettrone non accopiato del nitrosile può essere studiato tramite una tecnica chiamata EPR (Electron Paramagnetic Resonance) o ESR (Electron Spin Resonance). Esperimenti di questo tipo hanno dimostrato che gli spettri EPR sono sensibili ai sostituenti sul radicale così come al suo ambiente vicino. Questo ha portato ad una stategia di marcatura dello spin per l'indagine delle strutture conformazionali delle macromolecole come le proteine. percià, la tecnica site-directed spin labeling (SDSL) è emersa come tecnica preziosa per mappare gli elementi della struttura secondaria, al livello del fold del frammento, in un vasto range di proteine, incluse quelle non riconducibile alla caratterizzazione strutturale usando le classiche tecniche strutturali, quali l'NMR e la cristallografia ai raggi X. Grazie di aver fatto presente la cosa.
Grazie di aver fatto presente la cosa.