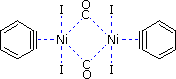quimico
2011-12-05 15:14
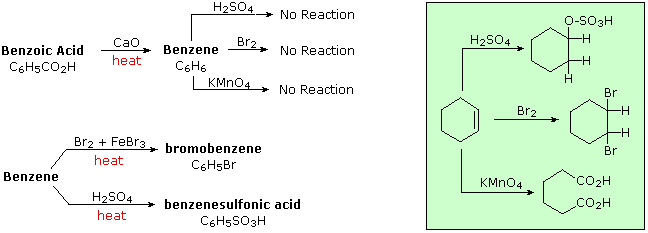
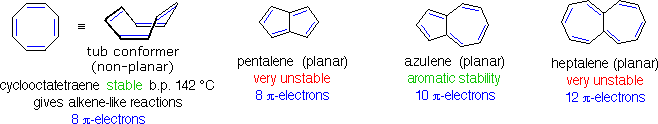
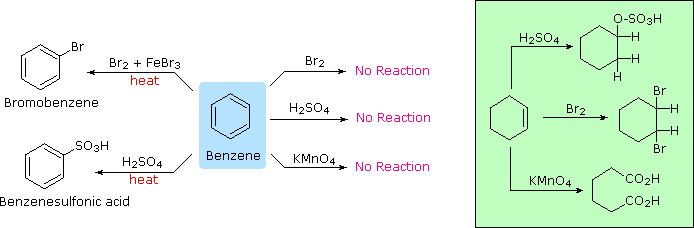
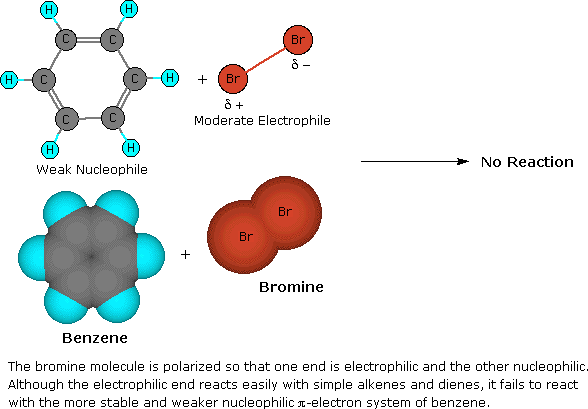
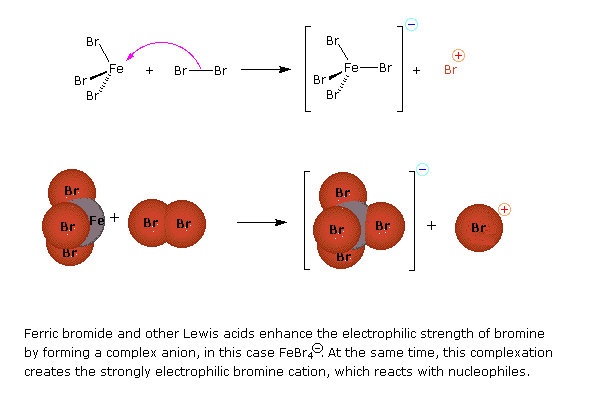
L'aggettivo "aromatico" viene usato dai chimici organici in un modo abbastanza diverso rispetto cui è normalmente applicato. Ha la sua origine nell'osservazione che alcuni sostanze naturali, quali la corteccia della cannella, le foglie di gaultheria del Canada, i bacelli di vaniglia e i semi di anice, contenevano composti profumati aventi proprietà comuni ma inaspettate. La corteccia della cannella, per esempio, produceva un composto dall'odore piacevole, di formula C9H8O, chiamato cinnamaldeide. A causa del rapporto basso tra idrogeno e carbonio in questo ed in altri composti aromatici (notate che il rapporto H:C in un alcano è >2), i chimici si aspettavano che le loro formule di struttura dovessero contenere un gran numero di doppi e tripli legami. Dato che i doppi legami sono facilmente rimossi da agenti ossidanti come potassio permanganato o ozono, e rapidamente addizionano bromo e cloro, queste reazioni vennero applicate a questi composti aromatici. Sorprendentemente, vennero ottenuti prodotti che apparivano ritenere molti dei doppi legami, e questi composti esibivano un elevato grado di stabilità chimica comparata a quella di alcheni e cicloalcheni noti (composti alifatici). Per trattamento con una soluzione calda di permanganato, la cinnamaldeide dava un composto stabile, cristallino, di formula C7H6O2, ora chiamato acido benzoico. Il rapporto H:C nell'acido benzoico è <1, suggerendo ancora la presenza di diversi doppi legami. L'acido benzoico fu infine convertito nello stabile idrocarburo benzene, C6H6, che anche esso si dimostrò non reattivo nei confronti delle comuni trasformazioni dei doppi legami, come mostrato sotto. Per paragone, anche le reazioni del cicloesene, un tipico alchene, con questi reagenti sono mostrate (riquadro verde). Fu acquisita come evidenza sperimentale per un vasto assortimento di composti, quelli che incorporavano questo nucleo eccezionalmente stabile iniziarono ad esser chiamati "aromatici".
 Se il benzene viene forzato a reagire aumentando la temperatura e/o aggiungendo un catalizzatore, esso subisce reazioni di sostituzione piuttosto che le reazioni di addizione che sono tipiche degli alcheni. Questo conferma ulteriormente la precedente asserzione che il nucleo a sei carboni del benzene è insolitamente stabile alla modificazione chimica. La contraddizione concetturale presentata dall'elevato grado di insaturazione (basso rapporto H:C) e l'elevata stabilità chimica per il benzene e i composti relativi rimase un enigma insoluto per molti anni. Alla fine, fu adottata la struttura ora accettata di un anello planari di carboni, di forma esagonale regolare, e l'eccezionale termodinamica e stabilità chimica di questo sistema fu attribuita alla stabilizzazione di risonanza di un triene ciclico coniugato.
Se il benzene viene forzato a reagire aumentando la temperatura e/o aggiungendo un catalizzatore, esso subisce reazioni di sostituzione piuttosto che le reazioni di addizione che sono tipiche degli alcheni. Questo conferma ulteriormente la precedente asserzione che il nucleo a sei carboni del benzene è insolitamente stabile alla modificazione chimica. La contraddizione concetturale presentata dall'elevato grado di insaturazione (basso rapporto H:C) e l'elevata stabilità chimica per il benzene e i composti relativi rimase un enigma insoluto per molti anni. Alla fine, fu adottata la struttura ora accettata di un anello planari di carboni, di forma esagonale regolare, e l'eccezionale termodinamica e stabilità chimica di questo sistema fu attribuita alla stabilizzazione di risonanza di un triene ciclico coniugato.
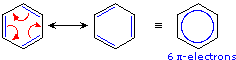 Qui, sono scritte due strutture elettroniche equivalenti strutturalmente ed energeticamente per un compsoto stabile, ma nessuna struttura singola fornisce un'accurata o anche un'adeguata rappresentazione della vera molecola. L'anelli a sei membri nel benzene è un esagono perfetto (tutti i legami carbonio-carbonio hanno un'identica lunghezza di 1.40 Å). Il cicloesatriene avrebbe lunghezze di legame alternate, i doppi legami sono più corti (1.34 Å) dei legami singoli (1.54 Å). Una rappresentazione alternativa per il benzene (un cerchio all'interno dell'esagono) enfatizza la delocalizzazione degli elettroni pi in questa molecola, ed ha il vantaggio di essere uno schema solo. In casi come questi, la delocalizzazione elettronica descritta dalla risonanza aumenta la stabilità delle molecole, e composti formati da tali molecole spesso mostrano un'eccezionale stabilità e relative proprietà.
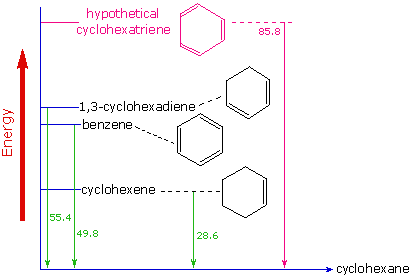
La prova dell'aumentata stabilità termodinamica del benzene è stata ottenuta da misure di calore rilasciato quando doppi legami in un anello a sei termini viene idrogenato (idrogeno è aggiunto cataliticamente) a dare il cicloesano come prodotto comune.
Nello schema seguente il cicloesano rappresenta un punto di riferimento a bassa energia. L'addizione dell'idrogeno al cicloesene produce il cicloesano e rilascia calore che ammonta a 28.6 kcal per mole. Se noi prendiamo questo valore per rappresentare il dispendio di energia per l'introduzione di un doppio legame in un anello a sei termini, dovremmo aspettarci che un cicloesadiene rilasci 57.2 kcal per mole per idrogenazione completa, e un 1,3,5-cicloesatriene rilasci 85.8 kcal per mole. Questi calori di idrogenazione dovrebbero riflettere la stabilità termodinamica relativa dei composti. In pratica, l'1,3-cicloesadiene è leggermente più stabile di quanti aspettato, di circa 2 kcal, presumibilmente a causa della coniugazione dei doppi legami. Il benzene, comunque, è staordinariamente di 36 kcal/mole più stabile di quanto aspettato. Questa sorta di aumento della stabilità è ora accettato come una caratteristica di tutti i composti aromatici.
Qui, sono scritte due strutture elettroniche equivalenti strutturalmente ed energeticamente per un compsoto stabile, ma nessuna struttura singola fornisce un'accurata o anche un'adeguata rappresentazione della vera molecola. L'anelli a sei membri nel benzene è un esagono perfetto (tutti i legami carbonio-carbonio hanno un'identica lunghezza di 1.40 Å). Il cicloesatriene avrebbe lunghezze di legame alternate, i doppi legami sono più corti (1.34 Å) dei legami singoli (1.54 Å). Una rappresentazione alternativa per il benzene (un cerchio all'interno dell'esagono) enfatizza la delocalizzazione degli elettroni pi in questa molecola, ed ha il vantaggio di essere uno schema solo. In casi come questi, la delocalizzazione elettronica descritta dalla risonanza aumenta la stabilità delle molecole, e composti formati da tali molecole spesso mostrano un'eccezionale stabilità e relative proprietà.
La prova dell'aumentata stabilità termodinamica del benzene è stata ottenuta da misure di calore rilasciato quando doppi legami in un anello a sei termini viene idrogenato (idrogeno è aggiunto cataliticamente) a dare il cicloesano come prodotto comune.
Nello schema seguente il cicloesano rappresenta un punto di riferimento a bassa energia. L'addizione dell'idrogeno al cicloesene produce il cicloesano e rilascia calore che ammonta a 28.6 kcal per mole. Se noi prendiamo questo valore per rappresentare il dispendio di energia per l'introduzione di un doppio legame in un anello a sei termini, dovremmo aspettarci che un cicloesadiene rilasci 57.2 kcal per mole per idrogenazione completa, e un 1,3,5-cicloesatriene rilasci 85.8 kcal per mole. Questi calori di idrogenazione dovrebbero riflettere la stabilità termodinamica relativa dei composti. In pratica, l'1,3-cicloesadiene è leggermente più stabile di quanti aspettato, di circa 2 kcal, presumibilmente a causa della coniugazione dei doppi legami. Il benzene, comunque, è staordinariamente di 36 kcal/mole più stabile di quanto aspettato. Questa sorta di aumento della stabilità è ora accettato come una caratteristica di tutti i composti aromatici.
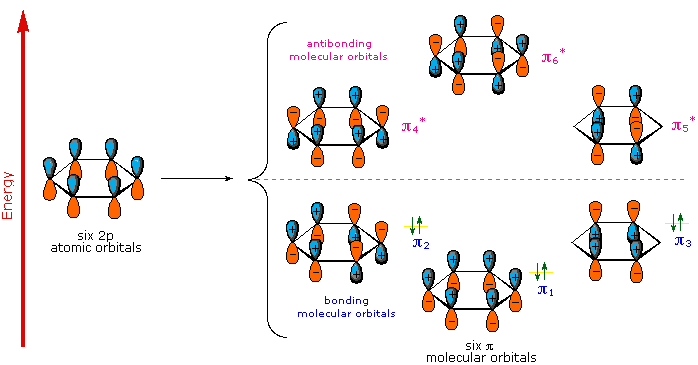
 Un descrizione MO del benzene fornisce un trattamento più soddisfacente e più generale della "aromaticità". Sappiamo che il benzene ha una struttura planare esagonale in cui tutti gli atomi di carbonio sono ibridizzati sp2, e tutt i legami carbonio-carbonio sono di eguale lunghezza. Come mostrato sotto, la rimanente disposizione ciclica dei sei orbitali p (uno su ogni carbonio) sovrappone generando sei orbitali molecolari, tre leganti e tre antileganti. I segni più e meno mostrati nello schema non rappresentano una carica elettrostatica, ma si riferiscono ai segni di fase nell'equazione che descrive questi orbitali (nello schema anche le fasi sono codificate da colori). Quando le gasi corrispondono, gli orbitali si sovrappongono a generare una regione comune di fase simile, con quegli orbitali che hanno la sovrapposizione maggiore (e.g. π1) essendo i più bassi in energia. I rimanenti elettroni di valenza del carbonio occupano quindi questi orbitali molecolari a coppie, che risultano in un set totalmente occupato (6 elettroni) di orbitali molecolari leganti. È questo set completamente pieno di orbitali leganti, o guscio chiuso, che dona all'anello del benzene la sua stabilità termodinamica e chimica, proprio come un ottetto completo di elettroni di valenza conferisce stabilità ai gas inerti.
Un descrizione MO del benzene fornisce un trattamento più soddisfacente e più generale della "aromaticità". Sappiamo che il benzene ha una struttura planare esagonale in cui tutti gli atomi di carbonio sono ibridizzati sp2, e tutt i legami carbonio-carbonio sono di eguale lunghezza. Come mostrato sotto, la rimanente disposizione ciclica dei sei orbitali p (uno su ogni carbonio) sovrappone generando sei orbitali molecolari, tre leganti e tre antileganti. I segni più e meno mostrati nello schema non rappresentano una carica elettrostatica, ma si riferiscono ai segni di fase nell'equazione che descrive questi orbitali (nello schema anche le fasi sono codificate da colori). Quando le gasi corrispondono, gli orbitali si sovrappongono a generare una regione comune di fase simile, con quegli orbitali che hanno la sovrapposizione maggiore (e.g. π1) essendo i più bassi in energia. I rimanenti elettroni di valenza del carbonio occupano quindi questi orbitali molecolari a coppie, che risultano in un set totalmente occupato (6 elettroni) di orbitali molecolari leganti. È questo set completamente pieno di orbitali leganti, o guscio chiuso, che dona all'anello del benzene la sua stabilità termodinamica e chimica, proprio come un ottetto completo di elettroni di valenza conferisce stabilità ai gas inerti.
I seguenti utenti ringraziano quimico per questo messaggio: Danny1212

 Molte altre reazioni di sostituzione del benzene sono state osservate, e le cinque più utili sono elencate sotto (clorurazione e bromurazione sono le più comuni reazioni di alogenazione). Dato che i reagenti e le condizioni impiegate in queste reazioni sono elettrofile, ci si riferisce comunemente a queste reazioni come sostituzione elettrofile aromatiche. I catalizzatori ed i co-reagenti servono a generare le forti specie elettrofile necessarie a compiere il passaggio iniziale di sostituzione.
Alogenazione: C6H6 + Cl2 o Br2 (a caldo, in presenza del catalizzatore FeCl3 o FeBr3) → C6H5Cl + HCl; elettrofilo: Cl+ o Br+
Nitrazione: C6H6 + HNO3 (a caldo, in presenza del catalizzatore H2SO4) → C6H5NO2 + H2O; elettrofilo: NO2+
Solfonazione: C6H6 + H2SO4 + SO3 (a caldo) → C6H5SO3H + H2O; elettrofilo: SO3H+
Alchilazione di Friedel-Crafts: C6H6 + R-Cl (a caldo in presenza del catalizzatore AlCl3) → C6H5-R + HCl; elettrofilo: R+
Acilazione di Friedel-Crafts: C6H6 + RCOCl (a caldo in presenza del catalizzatore AlCl3) → C6H6COR + HCl; elettrofilo: RCO+
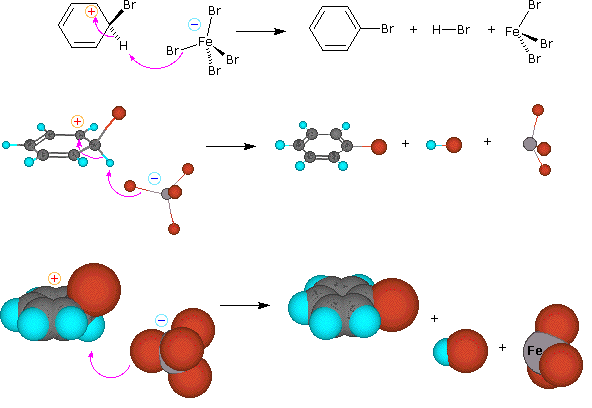
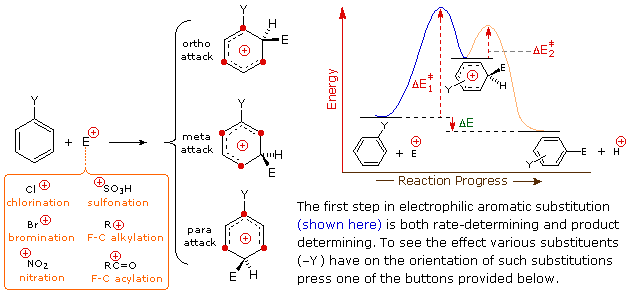
1. Un meccanismo per le reazioni di sostituzione elettrofila del benzene
Per queste reazioni di sostituzione elettrofila è stato proposto un meccanismo a due passaggi. Nel primo, lento o rate-determining, passaggio l'elettrofilo forma un legame sigma con l'anello benzenico, generando un intermedio benzenonio positivamente carico. Nel secondo, veloce passaggio, un protone è rimosso da quest intermedio, portando ad un anello benzenico sostituito.
Le quattro illustrazioni sotto mostrano questo meccanismo per la reazione di bromurazione.
Molte altre reazioni di sostituzione del benzene sono state osservate, e le cinque più utili sono elencate sotto (clorurazione e bromurazione sono le più comuni reazioni di alogenazione). Dato che i reagenti e le condizioni impiegate in queste reazioni sono elettrofile, ci si riferisce comunemente a queste reazioni come sostituzione elettrofile aromatiche. I catalizzatori ed i co-reagenti servono a generare le forti specie elettrofile necessarie a compiere il passaggio iniziale di sostituzione.
Alogenazione: C6H6 + Cl2 o Br2 (a caldo, in presenza del catalizzatore FeCl3 o FeBr3) → C6H5Cl + HCl; elettrofilo: Cl+ o Br+
Nitrazione: C6H6 + HNO3 (a caldo, in presenza del catalizzatore H2SO4) → C6H5NO2 + H2O; elettrofilo: NO2+
Solfonazione: C6H6 + H2SO4 + SO3 (a caldo) → C6H5SO3H + H2O; elettrofilo: SO3H+
Alchilazione di Friedel-Crafts: C6H6 + R-Cl (a caldo in presenza del catalizzatore AlCl3) → C6H5-R + HCl; elettrofilo: R+
Acilazione di Friedel-Crafts: C6H6 + RCOCl (a caldo in presenza del catalizzatore AlCl3) → C6H6COR + HCl; elettrofilo: RCO+
1. Un meccanismo per le reazioni di sostituzione elettrofila del benzene
Per queste reazioni di sostituzione elettrofila è stato proposto un meccanismo a due passaggi. Nel primo, lento o rate-determining, passaggio l'elettrofilo forma un legame sigma con l'anello benzenico, generando un intermedio benzenonio positivamente carico. Nel secondo, veloce passaggio, un protone è rimosso da quest intermedio, portando ad un anello benzenico sostituito.
Le quattro illustrazioni sotto mostrano questo meccanismo per la reazione di bromurazione.
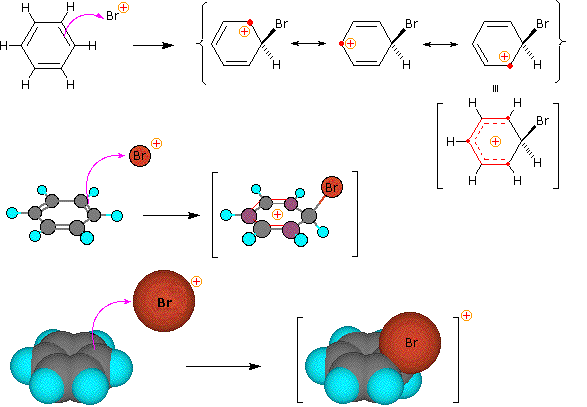
 Questo meccanismo per la sostituzione elettrofila aromatica dovrebbe essere considerato nel contesto assieme agli altri meccanismi che coinvolgono intermedi carbocationici. Questi includono reazioni SN1 ed E1 di alogenuri alchilici, e l'addizione di acidi di Brønsted degli alcheni.
Per riassumere, quando si formano intermedi carbocationici ci si può aspettare che essi reagiscano successivamente tramite uno o più dei seguenti modi:
1. Il catione può legarsi ad un nucleofilo a dare un prodotto di sostituzione o addizione.
2. Il catione può trasferire un protone ad una base, dando un prodotto con un doppio legame.
3. Il catione può riarrangiare ad un carbocatione più stabile, e quindi reagire tramite il modo #1 o #2.
Reazioni SN1 ed E1 sono i rispettivi esempi dei primi due modi di reazione. Il secondo passaggio delle reazioni di addizioni di alcheni procedono tramite il primo modo, e ognuna di queste tre reazioni può esibire riarrangiamento molecolare se si forma un iniziale carbocatione instabile. L'intermedio carbocationico nella sostituzione elettrofila aromatica (lo ione benzenonio) è stabilizzato tramite delocalizzazione di carica (risonanza) così non è soggetto a riarrangiamento. In principio esso potrebbe reagire in entrambi i modi 1 o 2, ma il vantaggio energetico del riformare un anello aromatico porta all'esclusiva reazione tramite il modo 2 (ovvero perdita del protone).
Questo meccanismo per la sostituzione elettrofila aromatica dovrebbe essere considerato nel contesto assieme agli altri meccanismi che coinvolgono intermedi carbocationici. Questi includono reazioni SN1 ed E1 di alogenuri alchilici, e l'addizione di acidi di Brønsted degli alcheni.
Per riassumere, quando si formano intermedi carbocationici ci si può aspettare che essi reagiscano successivamente tramite uno o più dei seguenti modi:
1. Il catione può legarsi ad un nucleofilo a dare un prodotto di sostituzione o addizione.
2. Il catione può trasferire un protone ad una base, dando un prodotto con un doppio legame.
3. Il catione può riarrangiare ad un carbocatione più stabile, e quindi reagire tramite il modo #1 o #2.
Reazioni SN1 ed E1 sono i rispettivi esempi dei primi due modi di reazione. Il secondo passaggio delle reazioni di addizioni di alcheni procedono tramite il primo modo, e ognuna di queste tre reazioni può esibire riarrangiamento molecolare se si forma un iniziale carbocatione instabile. L'intermedio carbocationico nella sostituzione elettrofila aromatica (lo ione benzenonio) è stabilizzato tramite delocalizzazione di carica (risonanza) così non è soggetto a riarrangiamento. In principio esso potrebbe reagire in entrambi i modi 1 o 2, ma il vantaggio energetico del riformare un anello aromatico porta all'esclusiva reazione tramite il modo 2 (ovvero perdita del protone).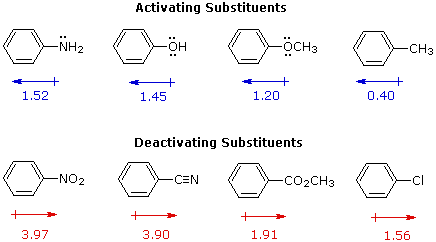
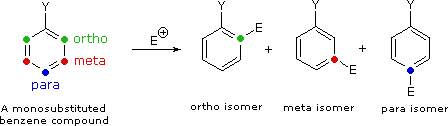
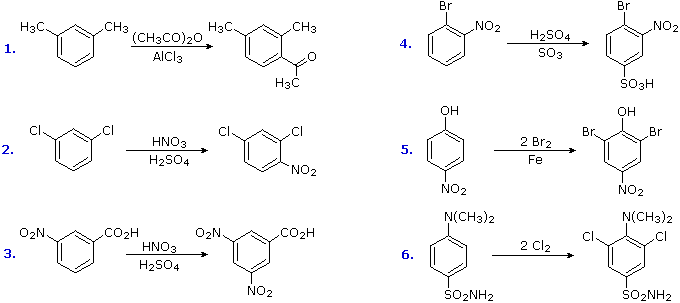
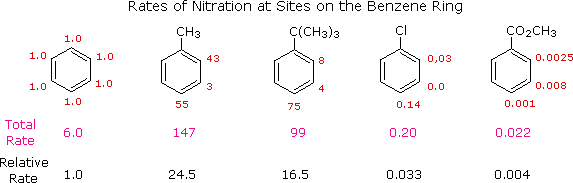 Da dati di questo tipo, è una semplice problema calcolare le proporzioni dei tre isomeri di sostituzione. Il toluene dà 58.5% di orto-nitrotoluene, 37% di para-nitrotoluene e solo il 4.5% dell'isomero meta. L'aumentato ingombro del gruppo tert-butile impedisce l'attacco delle posizioni orto, la miscela di prodotti totale è costituta per il 16% dal prodotto orto, 8% da quello meta e per il 75% dal prodotto para-nitro. Sebbene il clorobenzene siam molto meno reattivo del benzene, la velocità della orto e para sostituzione eccede di molto quella di meta sostituzione, dando una miscela di prodotto per il 30% orto e per il 70% para-nitroclorobenzene. Infine, l'estere benzoico porta predominantemente al prodotto meta-nitro (73%) accompagnato dagli isomeri orto (22%) e para (5%), come mostrato dalle velocità relative.
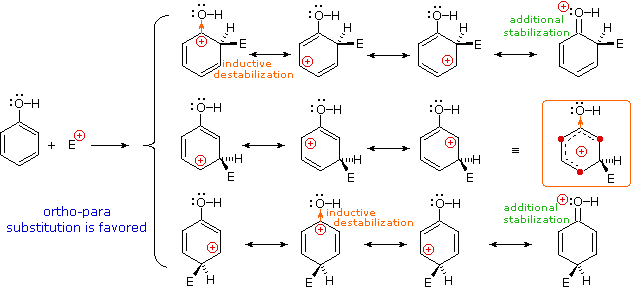
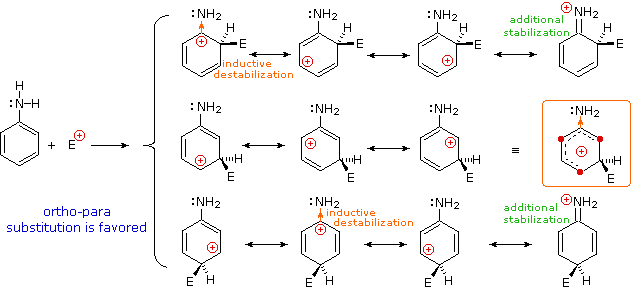
La maniera in cui specifici sostituenti influenzano l'orientazione della sostituzione elettrofila di un benzene è mostrata nel seguente schema. Come si nota dall'illustrazione, il passaggio che determina il prodotto nel meccanismo di sostituzione è il primo passaggio, che è anche il passaggio lento o rate determining step. Non è sorprendente, quindi, che c'è una grezza correlazione tra l'effetto che aumenta la velocità di un sostituente e la sua influenza che direziona il sito.
Da dati di questo tipo, è una semplice problema calcolare le proporzioni dei tre isomeri di sostituzione. Il toluene dà 58.5% di orto-nitrotoluene, 37% di para-nitrotoluene e solo il 4.5% dell'isomero meta. L'aumentato ingombro del gruppo tert-butile impedisce l'attacco delle posizioni orto, la miscela di prodotti totale è costituta per il 16% dal prodotto orto, 8% da quello meta e per il 75% dal prodotto para-nitro. Sebbene il clorobenzene siam molto meno reattivo del benzene, la velocità della orto e para sostituzione eccede di molto quella di meta sostituzione, dando una miscela di prodotto per il 30% orto e per il 70% para-nitroclorobenzene. Infine, l'estere benzoico porta predominantemente al prodotto meta-nitro (73%) accompagnato dagli isomeri orto (22%) e para (5%), come mostrato dalle velocità relative.
La maniera in cui specifici sostituenti influenzano l'orientazione della sostituzione elettrofila di un benzene è mostrata nel seguente schema. Come si nota dall'illustrazione, il passaggio che determina il prodotto nel meccanismo di sostituzione è il primo passaggio, che è anche il passaggio lento o rate determining step. Non è sorprendente, quindi, che c'è una grezza correlazione tra l'effetto che aumenta la velocità di un sostituente e la sua influenza che direziona il sito.
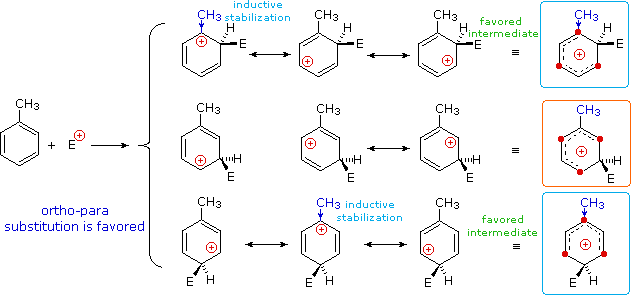
 Nel caso di sostituenti alchilici, la stabilizzazione di carica è la più grande quando il gruppo alchilico è legato ad uno dei carboni positivamente carichi dell'intermedio benzenonio. Questo avviene solo per l'attacco in orto e para, così tali sostituenti favorisocno la formazione di questi prodotti. È interessante notare che i sostituenti alchilici primari, specialmente metile, forniscono una stabilizzazione maggiore dell'adiacente carica rispetto a quanto fanno gruppi più sostituiti (notate la reattività maggiore del toluene comparata al tert-butilbenzene).
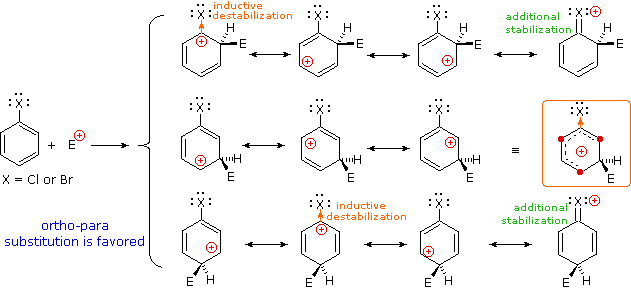
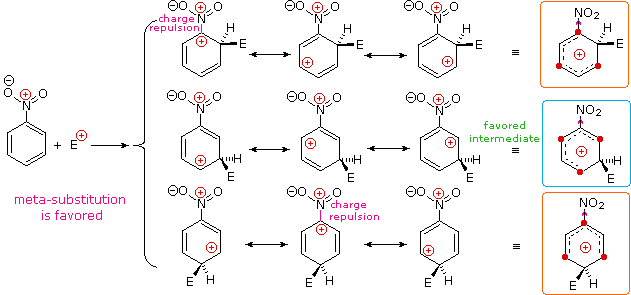
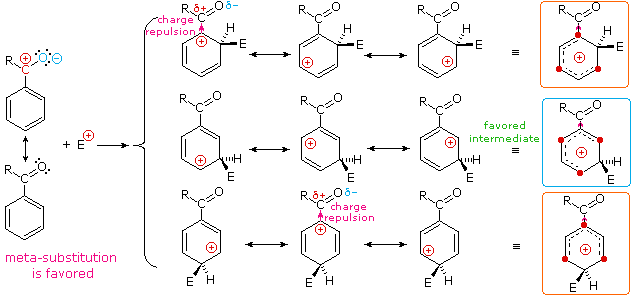
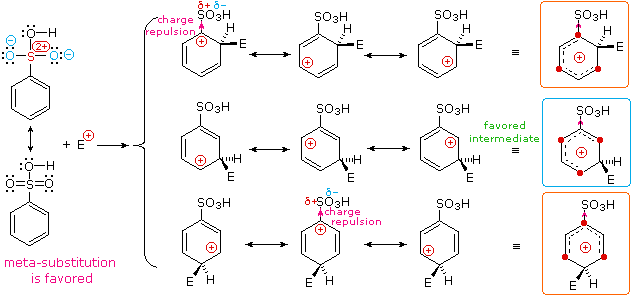
I sostituenti nitro (NO2), acido solfonico (SO3H) w carbonile (C=O) hanno una carica positiva totale o parziale sull'atomo legato all'anello aromatico. Strutture in cui cariche di questo tipo sono vicine l'una all'altra sono destabilizzate a causa della repulsione di carica, così questi sostituenti inibiscono la sostituzione in orto e para di più che la sostituzione in meta. Di conseguenza, i prodotti meta predominano quando la sostituzione elettrofila è forzata ad avvenire.
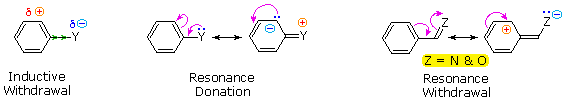
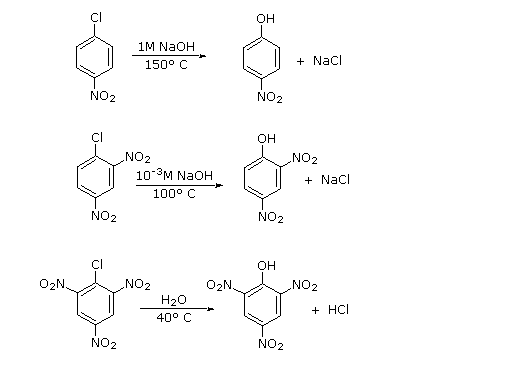
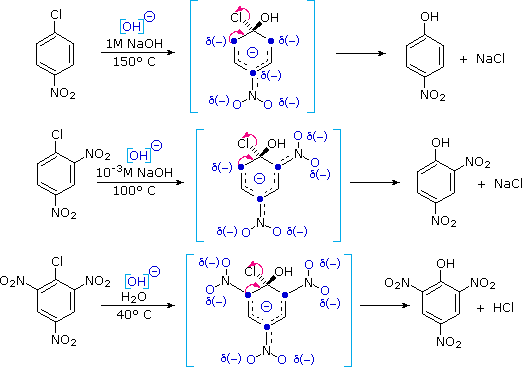
Sostituenti alogeno (X), OR e NR2 tutti esercitano un effetto induttivo destabilizzante sulle carice positive adiacenti, a causa dell'elevata elettronegatività degli atomi dei sostituenti. Da sé, questo dovrebbe favorire la sostituzione in meta; comunque, questi atomi dei sostituenti hanno tutti coppie di elettroni di valenza non leganti che servono a stabilizzare una carica positiva adiacente tramite legame pi, con una risultante delocalizzazione di carica. Di conseguenza, tutti questi sostituenti dirigono la sostituzione verso le posizioni orto e para. L'equilibrio tra risucchio elettronico induttivo e coniugazione p-π è tale che i sostituenti azotati ed ossigenati hanno un'influenza stabilizzante totale sull'intermedio benzenonio ed aumentano la velocità di sostituzione in modo marcato; invece i sostituenti alogenati hanno un'influenza destabilizzante totale.
Nel caso di sostituenti alchilici, la stabilizzazione di carica è la più grande quando il gruppo alchilico è legato ad uno dei carboni positivamente carichi dell'intermedio benzenonio. Questo avviene solo per l'attacco in orto e para, così tali sostituenti favorisocno la formazione di questi prodotti. È interessante notare che i sostituenti alchilici primari, specialmente metile, forniscono una stabilizzazione maggiore dell'adiacente carica rispetto a quanto fanno gruppi più sostituiti (notate la reattività maggiore del toluene comparata al tert-butilbenzene).
I sostituenti nitro (NO2), acido solfonico (SO3H) w carbonile (C=O) hanno una carica positiva totale o parziale sull'atomo legato all'anello aromatico. Strutture in cui cariche di questo tipo sono vicine l'una all'altra sono destabilizzate a causa della repulsione di carica, così questi sostituenti inibiscono la sostituzione in orto e para di più che la sostituzione in meta. Di conseguenza, i prodotti meta predominano quando la sostituzione elettrofila è forzata ad avvenire.
Sostituenti alogeno (X), OR e NR2 tutti esercitano un effetto induttivo destabilizzante sulle carice positive adiacenti, a causa dell'elevata elettronegatività degli atomi dei sostituenti. Da sé, questo dovrebbe favorire la sostituzione in meta; comunque, questi atomi dei sostituenti hanno tutti coppie di elettroni di valenza non leganti che servono a stabilizzare una carica positiva adiacente tramite legame pi, con una risultante delocalizzazione di carica. Di conseguenza, tutti questi sostituenti dirigono la sostituzione verso le posizioni orto e para. L'equilibrio tra risucchio elettronico induttivo e coniugazione p-π è tale che i sostituenti azotati ed ossigenati hanno un'influenza stabilizzante totale sull'intermedio benzenonio ed aumentano la velocità di sostituzione in modo marcato; invece i sostituenti alogenati hanno un'influenza destabilizzante totale.
 B. Riduzione di nitro gruppi e aril chetoni
Le reazioni di nitrazione elettrofila e di acilazione di Friedel-Crafts introducono sostituenti disattivanti, meta orientanti sull'anello aromatico. Gli atomi inseriti sono in uno stato di ossidazione elevati, e la loro riduzione converte queste funzioni elettron attratrici in gruppi elettron donatori amino ed alchile. La riduzione è facilmente ottenuta o tramite idrogenazione catalitica (H2 + catalizzatore), o tramite metalli riducenti in acido. Esempi di queste riduzioni sono mostrate qui; l'equazione 6 dimostra la simultanea riduzione di entrambe le funzioni. Notate che il prodotto butilbenzene nell'equazione 4 non può essere generato tramite diretta alchilazione di Friedel-Crafts a causa del riarrangiamento carbocationico. Lo zinco usato nelle riduzioni di chetoni, come nel caso 5, è di solito attivato legandolo con il mercurio (amalgamazione).
B. Riduzione di nitro gruppi e aril chetoni
Le reazioni di nitrazione elettrofila e di acilazione di Friedel-Crafts introducono sostituenti disattivanti, meta orientanti sull'anello aromatico. Gli atomi inseriti sono in uno stato di ossidazione elevati, e la loro riduzione converte queste funzioni elettron attratrici in gruppi elettron donatori amino ed alchile. La riduzione è facilmente ottenuta o tramite idrogenazione catalitica (H2 + catalizzatore), o tramite metalli riducenti in acido. Esempi di queste riduzioni sono mostrate qui; l'equazione 6 dimostra la simultanea riduzione di entrambe le funzioni. Notate che il prodotto butilbenzene nell'equazione 4 non può essere generato tramite diretta alchilazione di Friedel-Crafts a causa del riarrangiamento carbocationico. Lo zinco usato nelle riduzioni di chetoni, come nel caso 5, è di solito attivato legandolo con il mercurio (amalgamazione).
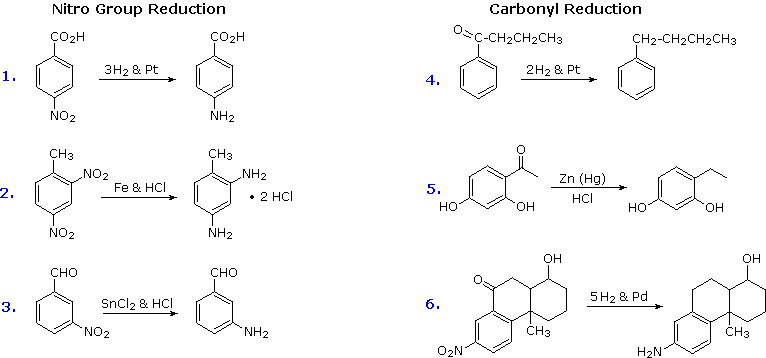 Diversi metodi alternativi per la riduzione di nitro gruppi ad amine sono noti. Questi includono zinco o stagno in acidi minerali diluiti, e sodio solfuro in soluzione di ammonio idrossido. Le procedure descritte sopra sono sufficienti per la maggior parte dei casi.
C. Conversione di alogeni in reagenti organometallici
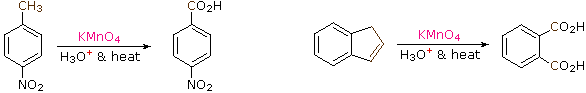
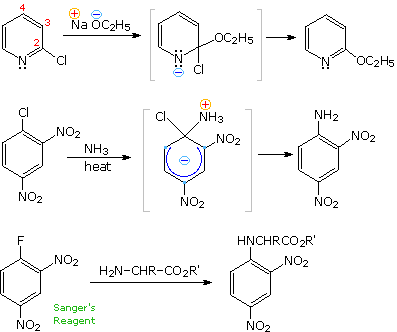
La reazione di alogenuri alchilici ed arilici con metalli reattivi (di solito Li & Mg) a dare reagenti nucleofili è stata notata. Questa fornisce un potente strumento per la conversione di sostituenti cloro, bromo o iodo in una varietà di altri gruppi. Molte reazioni di questi aril litio e reagenti di Grignard non saranno discusse qui, ma le equazioni seguenti forniscono esempi tipici di carbossilazione, protonazione e coupling di Gilman.
Diversi metodi alternativi per la riduzione di nitro gruppi ad amine sono noti. Questi includono zinco o stagno in acidi minerali diluiti, e sodio solfuro in soluzione di ammonio idrossido. Le procedure descritte sopra sono sufficienti per la maggior parte dei casi.
C. Conversione di alogeni in reagenti organometallici
La reazione di alogenuri alchilici ed arilici con metalli reattivi (di solito Li & Mg) a dare reagenti nucleofili è stata notata. Questa fornisce un potente strumento per la conversione di sostituenti cloro, bromo o iodo in una varietà di altri gruppi. Molte reazioni di questi aril litio e reagenti di Grignard non saranno discusse qui, ma le equazioni seguenti forniscono esempi tipici di carbossilazione, protonazione e coupling di Gilman.
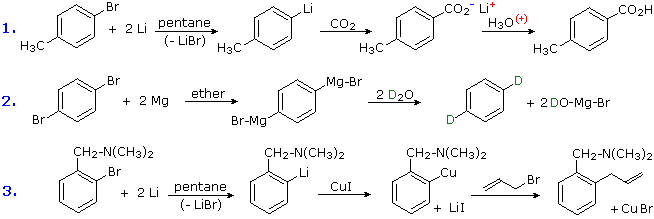 Le reazioni di scambio metallo-alogeno avvengono a bassa temperatura, e possono essere usate per introdurre lo iodio in posizioni designate. In questo esempo si deve aver cura di mantenere la temperatura bassa, perché per riscaldamento avviene eliminazione ad intermedio arinico.
Le reazioni di scambio metallo-alogeno avvengono a bassa temperatura, e possono essere usate per introdurre lo iodio in posizioni designate. In questo esempo si deve aver cura di mantenere la temperatura bassa, perché per riscaldamento avviene eliminazione ad intermedio arinico.
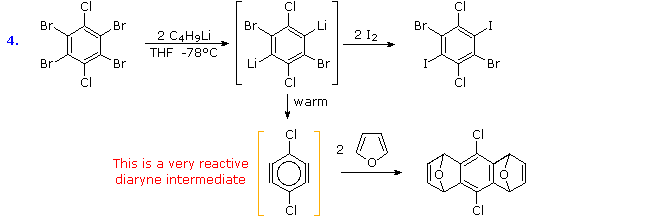 D. Idrolisi di acidi solfonici
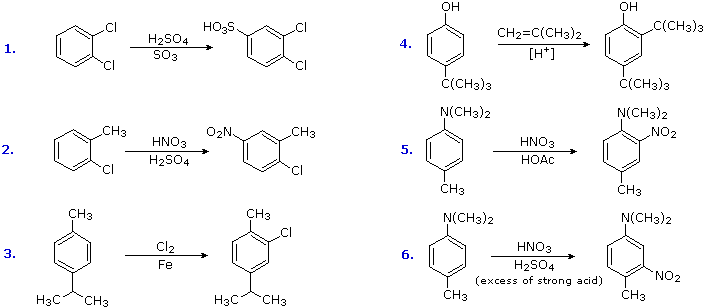
La potenziale reversibilità della reazione di solfonazione aromatica è un aspetto interessante di tale reazione. La seguente equazione illustra come questa caratteristica degli acidi solfonici può essere usata per preparare il 3-bromo derivato dell'orto-xilene. La bromurazione diretta dovrebbe dare il derivato 4-bromo.
D. Idrolisi di acidi solfonici
La potenziale reversibilità della reazione di solfonazione aromatica è un aspetto interessante di tale reazione. La seguente equazione illustra come questa caratteristica degli acidi solfonici può essere usata per preparare il 3-bromo derivato dell'orto-xilene. La bromurazione diretta dovrebbe dare il derivato 4-bromo.
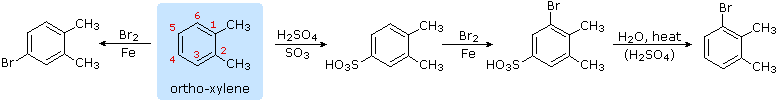 E. Modificando l'influenza dei forti gruppi attivanti
I sostituenti attivanti ed orto/para orientanti più forti sono i gruppi amino (-NH2) ed idrossile (-OH). La nitrazione diretta del fenolo (idrossibenzene) tramite acido nitrico diluito porta a fenoli nitrati con rese modeste e ad una considerevole decomposizione ossidativa a materiali catramosi; l'anilina (aminobenzene) è per la maggior parte distrutta. La bromurazione di entrambi è difficile da controllare, con formazione rapida di prodotti di- e tribromo. A causa della loro elevata reattività nucleofila, l'anilina ed il fenolo subiscono reazioni di sostituzione con iodio, un alogeno che è normalmente non reattivo verso i derivati del benzene. Il reagente misto iodio cloruro (ICl) fornisce una funzione iodio più elettrofila, ed è efficace nella iodurazione di anelli aromatici aventi sostituenti meno fortementi attivanti.
C6H5–NH2 + I2 + NaHCO3 → p-I-C6H4–NH2 + NaI + CO2 + H2O
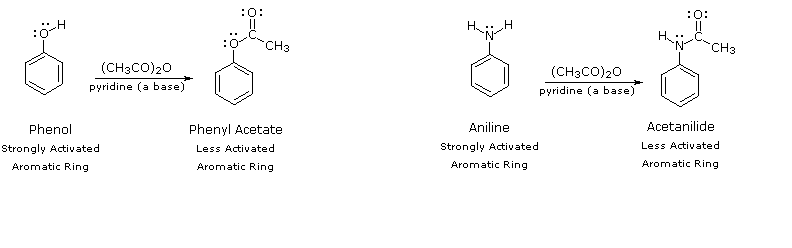
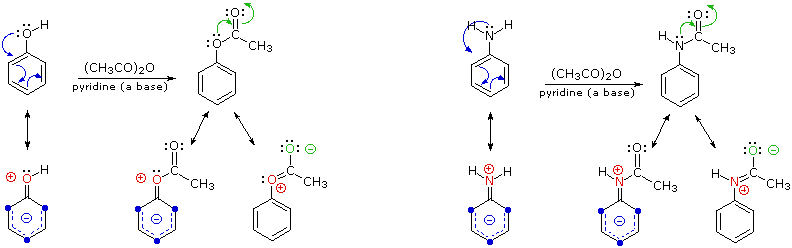
Acetilando il sostituente eteroatomico sul fenolo e sull'anilina, la sua influenza attivante può essere sostanzialmente attenuata. Per esempio, l'acetilazione dell'anilina porta alla acetanilide (primo passaggio nella equazione seguente), che subisce nitrazione a bassa temperatura, portando al prodotto para-nitro con resa elevata. Il gruppo modificante acetile può essere quindi rimosso tramite idrolisi acido-catalizzata (ultimo passaggio), a dare la para-nitroanilina. Sebbene l'influenza attivante del gruppo amino sia stata ridotta tramite questa procedura, il derivato acetile rimane un gruppo attivante ed orto/para orientante.
C6H5–NH2 + (CH3CO)2O (in piridina, base) → C6H5–NHCOCH3 + HNO3 (a 5 °C) → p-O2N–C6H4–NHCOCH3 + H3O+ (a caldo) → p-O2N–C6H4–NH2
Lo schema seguente illustra come il gruppo acetile agisca attenuando il carattere elettron donatore totale dell'ossigeno e dell'azoto. La coppia di elettroni di valenza non leganti che sono responsabili dell'elevata reattività di questi composti (frecce blu) è deviata verso l'adiacente gruppo carbonile (frecce verdi). Comunque, l'influenza totale del sostituente modificato è ancora attivante ed orto/para orientante.
E. Modificando l'influenza dei forti gruppi attivanti
I sostituenti attivanti ed orto/para orientanti più forti sono i gruppi amino (-NH2) ed idrossile (-OH). La nitrazione diretta del fenolo (idrossibenzene) tramite acido nitrico diluito porta a fenoli nitrati con rese modeste e ad una considerevole decomposizione ossidativa a materiali catramosi; l'anilina (aminobenzene) è per la maggior parte distrutta. La bromurazione di entrambi è difficile da controllare, con formazione rapida di prodotti di- e tribromo. A causa della loro elevata reattività nucleofila, l'anilina ed il fenolo subiscono reazioni di sostituzione con iodio, un alogeno che è normalmente non reattivo verso i derivati del benzene. Il reagente misto iodio cloruro (ICl) fornisce una funzione iodio più elettrofila, ed è efficace nella iodurazione di anelli aromatici aventi sostituenti meno fortementi attivanti.
C6H5–NH2 + I2 + NaHCO3 → p-I-C6H4–NH2 + NaI + CO2 + H2O
Acetilando il sostituente eteroatomico sul fenolo e sull'anilina, la sua influenza attivante può essere sostanzialmente attenuata. Per esempio, l'acetilazione dell'anilina porta alla acetanilide (primo passaggio nella equazione seguente), che subisce nitrazione a bassa temperatura, portando al prodotto para-nitro con resa elevata. Il gruppo modificante acetile può essere quindi rimosso tramite idrolisi acido-catalizzata (ultimo passaggio), a dare la para-nitroanilina. Sebbene l'influenza attivante del gruppo amino sia stata ridotta tramite questa procedura, il derivato acetile rimane un gruppo attivante ed orto/para orientante.
C6H5–NH2 + (CH3CO)2O (in piridina, base) → C6H5–NHCOCH3 + HNO3 (a 5 °C) → p-O2N–C6H4–NHCOCH3 + H3O+ (a caldo) → p-O2N–C6H4–NH2
Lo schema seguente illustra come il gruppo acetile agisca attenuando il carattere elettron donatore totale dell'ossigeno e dell'azoto. La coppia di elettroni di valenza non leganti che sono responsabili dell'elevata reattività di questi composti (frecce blu) è deviata verso l'adiacente gruppo carbonile (frecce verdi). Comunque, l'influenza totale del sostituente modificato è ancora attivante ed orto/para orientante.
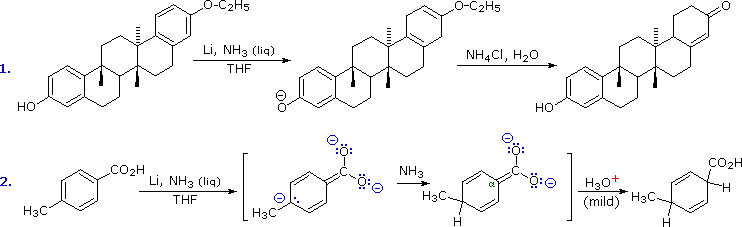
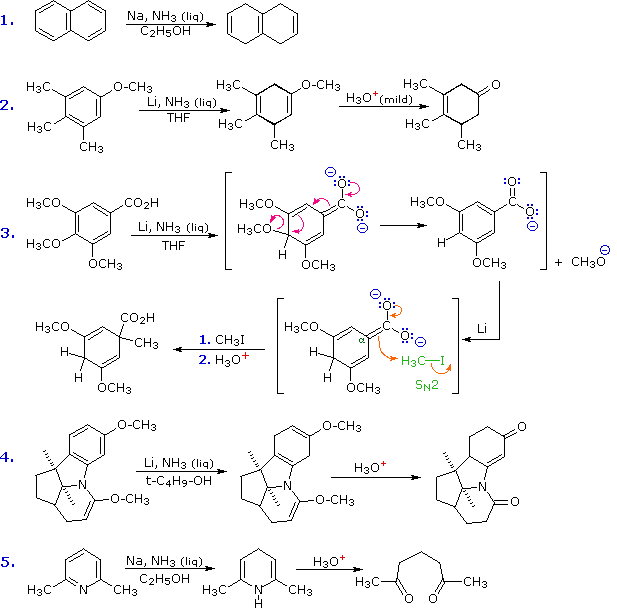
 Quando sono presenti dei sostituenti, essi possono influenzare la regioselettività della riduzione di Birch. Il prodotto è determinato dal site della prima protonazione, dato che la seconda protonazione è quasi sempre opposta (in para) rispetto alla prima. Sostituenti elettron donatori come gruppi eterei o alchilici favoriscono la protonazione al sito non occupato in orto al sostituente; invece sostituenti elettron attrattori cmoe un carbossile favoriscono la protonazione in para. L'influenza di un gruppo carbossile domina gli anelli poli sostituiti, ed i gruppo alcossi hanno una influenza orientante maggiore dei sostituenti alchilici. Un gruppo ossi anione, come nel caso della base coniugata del fenolo, previene che avvenga la riduzione. Due esempi di tali riduzioni di Birch sono mostrate sotto. Sebbene la molecola substrato nella prima reazione possa apparire davvero complessa, è essenzialmente una struttura rigida con un anello benzenico ad ogni estremità. La funzione fenolica sull'anello più a sinistra diventa un anione fenolato o fenossido nelle condizioni della riduzione, e non reagisce ulteriormente. L'anello aromatico più a destra è un etere, e si riduce come previsto. L'acido carbossilico nel secondo esempio è immediatamente convertito nella sua base coniugata. Sebbene questo anione carbossilato sia caricato negativamente, ha ancora un atomo di carbonio elettrofilo che agisce stabilizzando una adiacente carica negativa come mostrato. Dopo protonazione del carbanione in para tramite ammoniaca, il dianione carbossilato rimane intatto sino a quando è doppiamente protonato da un acido forte, come NH4+ o H3O+.
Quando sono presenti dei sostituenti, essi possono influenzare la regioselettività della riduzione di Birch. Il prodotto è determinato dal site della prima protonazione, dato che la seconda protonazione è quasi sempre opposta (in para) rispetto alla prima. Sostituenti elettron donatori come gruppi eterei o alchilici favoriscono la protonazione al sito non occupato in orto al sostituente; invece sostituenti elettron attrattori cmoe un carbossile favoriscono la protonazione in para. L'influenza di un gruppo carbossile domina gli anelli poli sostituiti, ed i gruppo alcossi hanno una influenza orientante maggiore dei sostituenti alchilici. Un gruppo ossi anione, come nel caso della base coniugata del fenolo, previene che avvenga la riduzione. Due esempi di tali riduzioni di Birch sono mostrate sotto. Sebbene la molecola substrato nella prima reazione possa apparire davvero complessa, è essenzialmente una struttura rigida con un anello benzenico ad ogni estremità. La funzione fenolica sull'anello più a sinistra diventa un anione fenolato o fenossido nelle condizioni della riduzione, e non reagisce ulteriormente. L'anello aromatico più a destra è un etere, e si riduce come previsto. L'acido carbossilico nel secondo esempio è immediatamente convertito nella sua base coniugata. Sebbene questo anione carbossilato sia caricato negativamente, ha ancora un atomo di carbonio elettrofilo che agisce stabilizzando una adiacente carica negativa come mostrato. Dopo protonazione del carbanione in para tramite ammoniaca, il dianione carbossilato rimane intatto sino a quando è doppiamente protonato da un acido forte, come NH4+ o H3O+.
 Ulteriori esempi delle riduzioni di Birch sono presentati nello schema seguente. La preferenza per la protonazione nei siti non sostituiti (nonostante siano presenti gruppi elettron attrattori), e per prodotti non coniugati è ancora illustrata nella prima reazione. Notate che i doppi legami isolati non sono ridotti a basse temperature in ammoniaca a riflusso (–33 °C). Le reazioni #2 & #4 illustrano una applicazione particolarmente utile della riduzione di Birch. Gi aril eteri sono ridotti ad 1,4-dieni, come si aspetta, ma uno dei doppi legami è un enol etere ed è rapidamente idrolizzato al chetone corrispondente. Se si usa una blanda catalisi acida, l'altro doppio legame rimane inalterato; un trattamento acido (o basico) più forte shifta questo doppio legame verso una posizione coniugata se il semplice shift di protone lo permette. La reazione #3 illustra di nuovo l'influenza regio-orientante di un gruppo carbossile, anche nella forma carbossilato. L'alfa anione è sufficientemente stabile che induce una reazione di eliminazione (primo stadio) e subito dopo rigenerazione può essere alchilata da un alogenuro alchilico reattivo (secondo stadio). L'ultimo esempio mostra la riduzione di Birch della piridina ad una bis-enammina, la cui idrolisi porta ad un dichetone.
Ulteriori esempi delle riduzioni di Birch sono presentati nello schema seguente. La preferenza per la protonazione nei siti non sostituiti (nonostante siano presenti gruppi elettron attrattori), e per prodotti non coniugati è ancora illustrata nella prima reazione. Notate che i doppi legami isolati non sono ridotti a basse temperature in ammoniaca a riflusso (–33 °C). Le reazioni #2 & #4 illustrano una applicazione particolarmente utile della riduzione di Birch. Gi aril eteri sono ridotti ad 1,4-dieni, come si aspetta, ma uno dei doppi legami è un enol etere ed è rapidamente idrolizzato al chetone corrispondente. Se si usa una blanda catalisi acida, l'altro doppio legame rimane inalterato; un trattamento acido (o basico) più forte shifta questo doppio legame verso una posizione coniugata se il semplice shift di protone lo permette. La reazione #3 illustra di nuovo l'influenza regio-orientante di un gruppo carbossile, anche nella forma carbossilato. L'alfa anione è sufficientemente stabile che induce una reazione di eliminazione (primo stadio) e subito dopo rigenerazione può essere alchilata da un alogenuro alchilico reattivo (secondo stadio). L'ultimo esempio mostra la riduzione di Birch della piridina ad una bis-enammina, la cui idrolisi porta ad un dichetone.
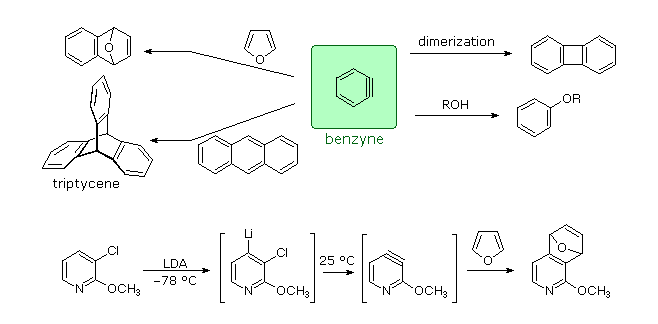
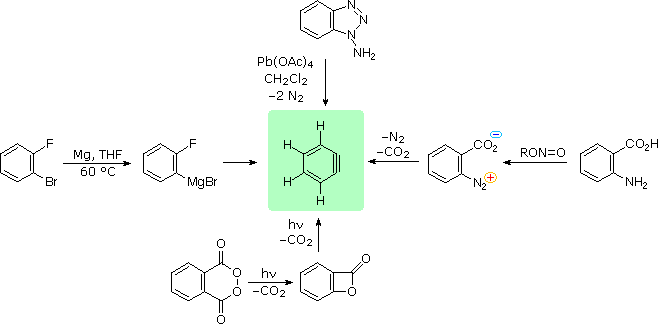 Dato che il benzino (e gli altri arini) è un potente dienofilo, molte delle sue reazioni di addizione sono cicloaddizioni. Notate l'analogo piridinico del benzino nella equazione in basso.
Dato che il benzino (e gli altri arini) è un potente dienofilo, molte delle sue reazioni di addizione sono cicloaddizioni. Notate l'analogo piridinico del benzino nella equazione in basso.
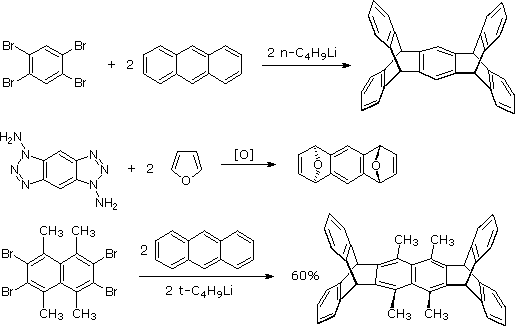
 Arini e diarini sono stati utilizzati nella sintesi di molecole aromatiche multi pontate chiamate tripticeni. Alcuni esempi sono riportati nello schema seguente.
Arini e diarini sono stati utilizzati nella sintesi di molecole aromatiche multi pontate chiamate tripticeni. Alcuni esempi sono riportati nello schema seguente.
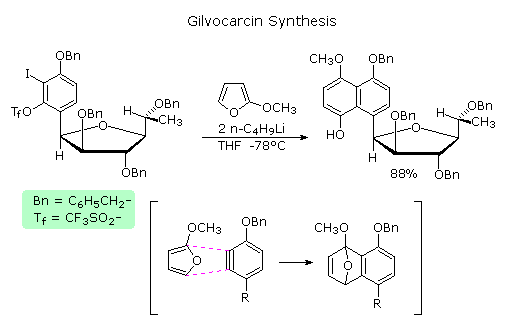
 Sotto è riportata una applicazione sintetica della cicloaddizione di un arino al fine di ottenere un prodotto naturale. In questo caso l'intermedio arino si cicloaddiziona al furano sostituito in un modo altamente regioselettivo, come mostrato tra parentesi. L'addotto iniziale subisce quindi una rapida apertura d'anello eliminativa a derivato naftalenico.
Sotto è riportata una applicazione sintetica della cicloaddizione di un arino al fine di ottenere un prodotto naturale. In questo caso l'intermedio arino si cicloaddiziona al furano sostituito in un modo altamente regioselettivo, come mostrato tra parentesi. L'addotto iniziale subisce quindi una rapida apertura d'anello eliminativa a derivato naftalenico.
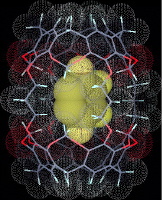
 Sebbene gli arini siano estremamente reattivi, è stato possibile esaminarli spettroscopicamente in raggi molecolari nell'alto vuoto, ed intrappolati in matrici vetrose inerti a temperatura molto basse. Come mostrato sotto, il benzino stesso è stato catturato in una gabbia molecolare, definita un carcerando.
Sebbene gli arini siano estremamente reattivi, è stato possibile esaminarli spettroscopicamente in raggi molecolari nell'alto vuoto, ed intrappolati in matrici vetrose inerti a temperatura molto basse. Come mostrato sotto, il benzino stesso è stato catturato in una gabbia molecolare, definita un carcerando.
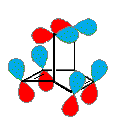
 Il benzino forma anche complessi π stabili con alcuni metalli di transizione, essendone un esempio il complesso di Ni disegnato sotto.
Il benzino forma anche complessi π stabili con alcuni metalli di transizione, essendone un esempio il complesso di Ni disegnato sotto.