6. Glicosidi
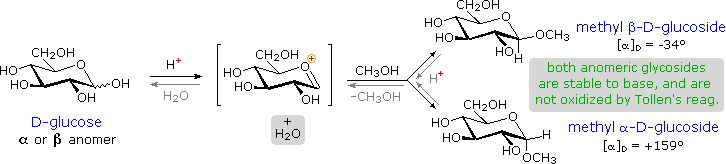
I derivati acetalici formati quando un monosaccaride reagisce con un alcole in presenza di un catalizzatore acido sono chiamati glicosidi. Questa reazione è illustrata per il glucosio ed il metanolo nello schema sotto. Nel chiamare i glicosidi, il suffisso "osio" del nome dello zucchero viene sostituito da "oside", ed il nome del gruppo alcolico è messo per primo. Dato che è generalmente vero per la maggior parte degli acetali, la formazione di un glucoside comporta la perdita di un equivalente di acqua. Il dietere prodotto è stabile alle basi e agli ossidanti alcalini come il reagente di Tollen. Dato che una aldolizzazione acido catalizzata è reversibile, i glicosidi possono essere idrolizzati di nuovo ai loro alcoli e ai loro zuccheri componenti tramite acido acquoso.
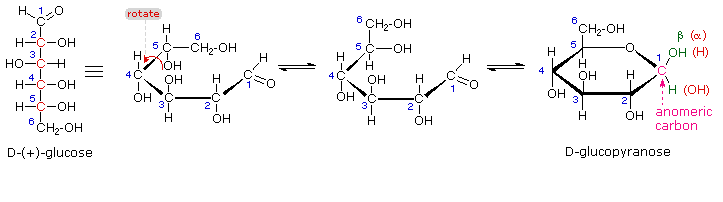
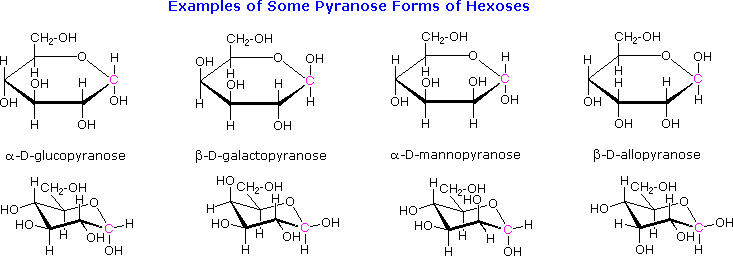
I metil glicosidi anomerici sono formati in un rapporto di equilibrio di 66% α contro il 34% β. Dalle strutture nello schema precedente, vediamo che gli anelli piranosidici preferiscono conformazioni a sedia in cui il maggior numero di sostituenti sono equatoriali. Nel caso del glucosio, i sostituenti sull'anomero β sono tutti equatoriali, mentre il sostituente al C-1 nell'anomero α cambia ad assiale. Dato che i sostituenti su anelli cicloesanici preferiscono una posizione equatoriale rispetto ad un assiale (il metossicicloesano è per il 75% equatoriale), la preferenza per la formazione dell'α-glicopiranoside è inaspettata, e ci si riferisce ad essa come effetto anomerico.
I glicosidi abbondano nei sistemi biologici. Attaccando una metà carboidrato ad una lipide o ad una struttura benzenoide, la solubilità e le altre proprietà del composto possono essere cambiate sostanzialmente. A causa dell'importante influenza modificante di tale derivatizzazione, numerosi sistemi enziamatici, noti come glicosidasi, si sono sviluppati per attacco e rimozione di zuccheri da alcoli, fenoli ed amine. I chimici si riferiscono alla componente carboidrato di glicosidi naturali con il termine glicone ed alal componente alcolica con il termine aglicone. Due esempi di glicosidi che si trovano in natura ed un esempio di un amino derivato sono mostrato sotto.
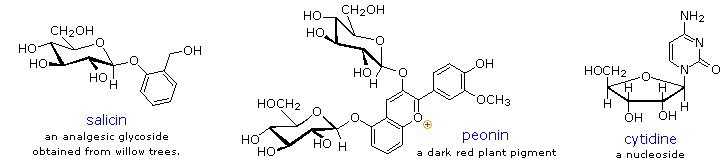
La salicina, uno dei pià antichi rimedi erboristici noto, fu il modello per l'analgesico sintetico aspirina. Una grossa classe di cationi ossonio aromatici, idrossilati chiamati antocianine forniscono i colori rosso, viola e blu di molti fiori, frutti ed alcuni vegetali.
La peonina è un esempio di questa classe di pigmenti naturali, che esibiscono una pronunciata dipendenza del colore dal pH. La metà ossonio è solo stabile in ambienti acidi, ed il colore cambia o sparisce quando viene aggiunta una base. I cambiamenti complessi che avvengono quando il vino viene fermentato e conservato sono in parte associati con i glicosidi delle antocianine.
Infine, un amino derivato del ribosio, quale la citidina gioca importanti ruoli negli agenti biologici fosforilanti, nei coenzimi e nei materiali di traporto e conservazione dell'informazione.
7. Disaccaridi
Quando il componente alcolico di un glicoside è unito tramite una funzione idrossile su un altro monosaccaride, il composto viene chiamato disaccaride. Quattro esempi di disaccaridi composti di due unità di glucodio sono mostrati nel seguente schema. Gli anelli glicopiranosidici individuali sono etichettati A e B, ed il legame glicosidico è cerchiato in blu chiaro. Notate che il legame glicosidico può essere α, come nel maltosio e nel trealosio, o β come nel cellobiosio e nel gentiobiosio. L'idrolisi acido catalizzata di questi disaccaridi porta al glucosio come unico prodotto. L'idrolisi catalizzata da enzima è selettiva per uno specifico legame glicosidico, così un'α-glicosidasi scinde il maltosio ed il trealosio in glucosio, ma non scinde il cellobiosio o il gentiobiosio. Una β-glicosidasi ha l'attività opposta.
Al fine di disegnare una struttura rappresentativa per il cellobiosio, uno degli anelli glucopiranosidici deve essere ruotato di 180°, ma questa caratteristica è spesso omessa in favore della ritenzione della prospettiva solita per gli anelli individuali. Il legame tra gli anelli glicopiranosidici nel cellobiosio e nel maltosio è dal carbonio anomerico nell'anello A verso il gruppo idrossile al C-4 sull'anello B. Questo lascia il carbonio anomerico nell'anello B libero, così cellobiosio e maltosio possono entrambi assumere anomeri α e β a quel sito (la forma β è mostrata nello schema). Il gentiobiosio ha una connessione β-glicosidica, che ha origine al C-1 nell'anello A e termina al C-6 nell'anello B. Il suo anomero α è disegnato nello schema. Poiché il cellobiosio, il maltosio ed il gentiobiosio sono emiacetali essi sono tutti zuccheri riducenti (ossidati dal reagente di Tollen). Il trealosio, un disaccaride trovato in alcuni funghi, è un bis-acetale, ed è quindi uno zucchero non riducente. Una nomenclatura sistematica per i disaccaridi esiste, ma come gli esempi seguenti illustrano, questi nomi sono spesso prolissi.
Cellobiosio: 4-O-β-D-Glucopiranosil-D-glucosio (è disegnato l'anomero β)
Maltosio: 4-O-α-D-Glucopiranosil-D-glucosio (è disegnato l'anomero β)
Gentiobiosio: 6-O-β-D-Glucopiranosil-D-glucosio (è disegnato l'anomero α)
Trealosio: α-D-Glucopiranosil-α-D-glucopiranoside
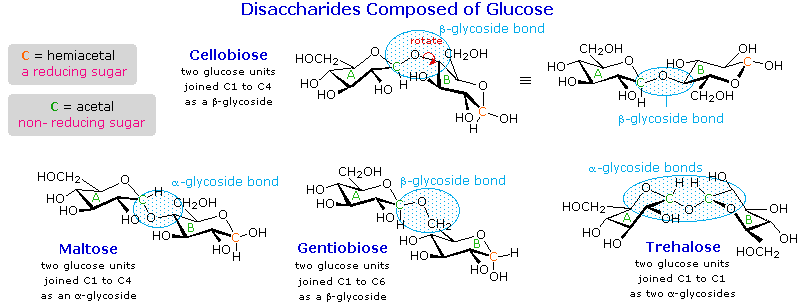
Sebbene tutti i disaccaridi mostrati qui siano formati da due anelli glucopiranosidici, le loro proprietà differiscono in modi interessanti.
Il maltosio, a volte chiamato zucchero di malto, deriva dall'idrolisi dell'amido. È circa 1/3 dolce quanto lo zucchero di canna (saccarosio), è facilmente digerito dagli umani, ed è fermentato dal lievito.
Il cellobiosio viene ottenuto tramite idrolisi della cellulosa. Non ha virtualmente alcun sapore, è indigeribile per gli umani, e non viene fermentato dal lievito. Alcuni batteri hanno enzimi β-glucosidasi che idrolizzano i legami glicosidici nel cellobiosio e nella cellulosa. La presenza di tali batteri nel tratto digerente di vacche e termiti permette a questi animali di usare la cellulosa come cibo.
Infine, può essere notato che il trealosio ha un sapore distintamente dolce, ma il gentiobiosio è amaro.
I disaccaridi formati da altri zuccheri sono noti, ma il glucosio è spesso uno dei componenti.
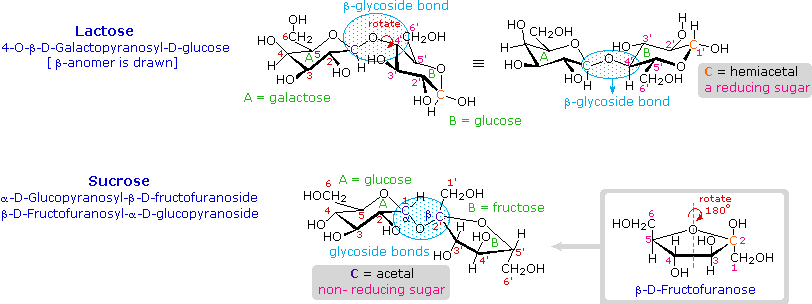
Due esempi importanti di tali disaccaridi misti è mostrato sopra.
Il lattosio, anche noto come zucchero del latte, è un composto galattosio-glucosio unito come β-glicoside. È uno zucchero riducente a causa della funzionale emiacetalica che rimane nella metà glucosio. Molti adulti, particolarmente quelli da regioni dove il latte non è alimento base della dieta, hanno una intolleranza metabolica per il lattosio. I neonati hanno un enzima digestivo che scinde il legame β-glicosidico nel lattosio, ma la produzione di questo enzima si ferma con lo svezzamento. Il formaggio è meno soggetto al problema dell'intolleranza al lattosio, dato che la maggior parte del lattosio viene rimosso con il siero.
Il saccarosio, o zucchero di canna, è il nostro agente dolcificante più comunemente usato. È uno zucchero non riducente composto da glucosio e fruttosio uniti al carbonio anomerico di ognuno tramite legami glicosidici (uno α ed uno β). Nella formula mostrata qui l'anello del fruttosio è stato rotato di 180° rispetto alla sua prospettiva convezionale.
 Perciò ho deciso di aprire qui in chimica organica tale discussione. Se volete spostarla magari avvisatemi, grazie
Perciò ho deciso di aprire qui in chimica organica tale discussione. Se volete spostarla magari avvisatemi, grazie  .
Iniziamo.
I carboidrati sono la classe più abbondante di composti organici trovati negli organismi viventi. Essi nascono come prodotti della fotosintesi, una condensazione riduttiva endotermica di diossido di carbonio che richiede luce come fonte di energia ed il pigmento clorofilla.
n CO2 + n H2O + energia → CnH2nOn + n O2
Come si nota qui, le formule di molti carboidrati possono essere scritte come idrati del carbonio, Cn(H2O)n, da qui il loro nome. I carboidrati sono la principale fonte dell'energia metabolica, sia per le piante che per gli animali che dipendono dalle piante per il nutrimento. A parte gli zucchieri e gli amidi che si rispondono a questo ruolo nutrizionale vitale, i carboidati servono anche come materiale strutturale (cellulosa), come componente dell'ATP composto adibito al trasposto energetico, come siti di riconoscimenti sulle superficie cellulare, e come uno dei tre componenti essenziali del DNA e dell'RNA.
I carboidrati sono chiamati saccaridi o, se essi sono relativamente piccoli, zuccheri. Diverse classificazioni dei carboidrati si sono dimostrate utili, e sono riassunte sotto.
Per quanto riguarda la complessità, esistono: carboidrati semplici (monosaccaridi) e carboidrati complessi (disaccaridi, oligosaccaridi
& polisaccaridi).
Per quanto riguarda la dimensione, abbiamo tetrosi (zuccheri a 4 atomi di carbonio), pentosi (zuccheri a 5 atomi di carbonio), esosi (zuccheri a 6 atomi di carbonio), eptosi (zuccheri a 7 atomi di carbonio), etc.
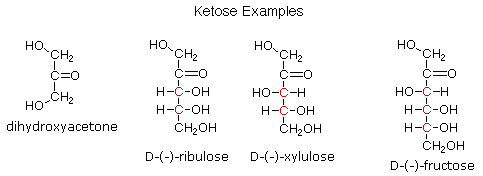
Per quanto riguarda la funzionalità C=O, i carboidrati si dividono in aldosi (zuccheri aventi una funzione aldeidica o un acetale equivalente) e chetosi (zuccheri aventi una funzione chetonica o un acetale equivalente).
Infine, per quanto concerne la reattività, gli zuccheri possono essere riducenti (ossidati dal reagente di Tollens o dai reagenti di Benedict o di Fehling) o non riducenti (non ossidati dal reagente di Tollens o dai reagenti di Benedict o di Fehling).
.
Iniziamo.
I carboidrati sono la classe più abbondante di composti organici trovati negli organismi viventi. Essi nascono come prodotti della fotosintesi, una condensazione riduttiva endotermica di diossido di carbonio che richiede luce come fonte di energia ed il pigmento clorofilla.
n CO2 + n H2O + energia → CnH2nOn + n O2
Come si nota qui, le formule di molti carboidrati possono essere scritte come idrati del carbonio, Cn(H2O)n, da qui il loro nome. I carboidrati sono la principale fonte dell'energia metabolica, sia per le piante che per gli animali che dipendono dalle piante per il nutrimento. A parte gli zucchieri e gli amidi che si rispondono a questo ruolo nutrizionale vitale, i carboidati servono anche come materiale strutturale (cellulosa), come componente dell'ATP composto adibito al trasposto energetico, come siti di riconoscimenti sulle superficie cellulare, e come uno dei tre componenti essenziali del DNA e dell'RNA.
I carboidrati sono chiamati saccaridi o, se essi sono relativamente piccoli, zuccheri. Diverse classificazioni dei carboidrati si sono dimostrate utili, e sono riassunte sotto.
Per quanto riguarda la complessità, esistono: carboidrati semplici (monosaccaridi) e carboidrati complessi (disaccaridi, oligosaccaridi
& polisaccaridi).
Per quanto riguarda la dimensione, abbiamo tetrosi (zuccheri a 4 atomi di carbonio), pentosi (zuccheri a 5 atomi di carbonio), esosi (zuccheri a 6 atomi di carbonio), eptosi (zuccheri a 7 atomi di carbonio), etc.
Per quanto riguarda la funzionalità C=O, i carboidrati si dividono in aldosi (zuccheri aventi una funzione aldeidica o un acetale equivalente) e chetosi (zuccheri aventi una funzione chetonica o un acetale equivalente).
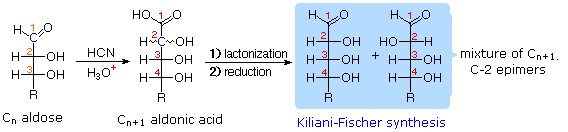
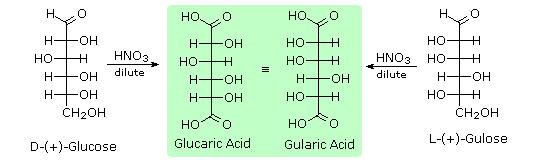
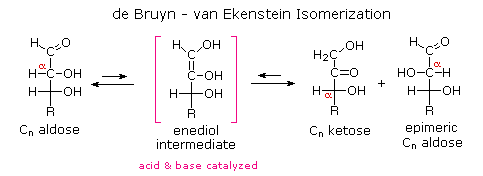
Infine, per quanto concerne la reattività, gli zuccheri possono essere riducenti (ossidati dal reagente di Tollens o dai reagenti di Benedict o di Fehling) o non riducenti (non ossidati dal reagente di Tollens o dai reagenti di Benedict o di Fehling).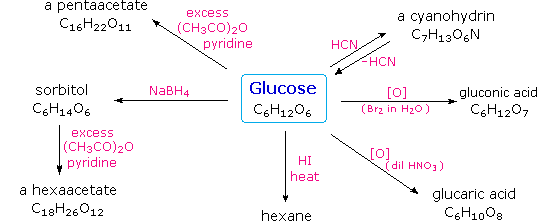 L'acido iodidrico a caldo (HI) fu spesso usato per rimuovere riduttivamente gruppi funzionali ossigenati da una molecola, e nel caso del glucosio questo trattamento portò all'esano (con resa bassa). Da ciò venne concluso che i sei carboni sono in una catena non ramificata. La presenza di un gruppo carbonilico (aldeide) fu dedotta dalla formazione di una cianoidrina, dalla sua riduzione ad esaalcole sorbitolo, anche chiamato glucitolo, e dalla blanda ossidazione ad acido monocarbossilico, acido glucuronico. L'ossidazione un po' più forte tramite acido nitrico diluito diede il diacido, acido glucarico, supportando la proposta di una catena a sei atomi di carbonio. I cinque ossigeni rimanenti nel glucosio, dopo che venne identificata l'aldeide, vennero ritenuti essere parte di un gruppo idrossile, dato che poteva esser fatto un derivato pentaacetato. Questi gruppi idrossile furono assegnati, uno per uno, agli ultimi cinque atomi di carbonio, perché gruppi idrossilici geminali sono normalmente instabili rispetto al composti carbonilico formato per perdita di acqua.
L'acido iodidrico a caldo (HI) fu spesso usato per rimuovere riduttivamente gruppi funzionali ossigenati da una molecola, e nel caso del glucosio questo trattamento portò all'esano (con resa bassa). Da ciò venne concluso che i sei carboni sono in una catena non ramificata. La presenza di un gruppo carbonilico (aldeide) fu dedotta dalla formazione di una cianoidrina, dalla sua riduzione ad esaalcole sorbitolo, anche chiamato glucitolo, e dalla blanda ossidazione ad acido monocarbossilico, acido glucuronico. L'ossidazione un po' più forte tramite acido nitrico diluito diede il diacido, acido glucarico, supportando la proposta di una catena a sei atomi di carbonio. I cinque ossigeni rimanenti nel glucosio, dopo che venne identificata l'aldeide, vennero ritenuti essere parte di un gruppo idrossile, dato che poteva esser fatto un derivato pentaacetato. Questi gruppi idrossile furono assegnati, uno per uno, agli ultimi cinque atomi di carbonio, perché gruppi idrossilici geminali sono normalmente instabili rispetto al composti carbonilico formato per perdita di acqua.
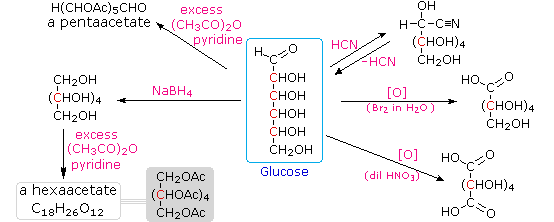 Sopra sono miostrati i prodotti proposti e la struttura generale del glucosio. I quattro atomi di carbonio centrali sono centri chirali e sono colorati in rosso.
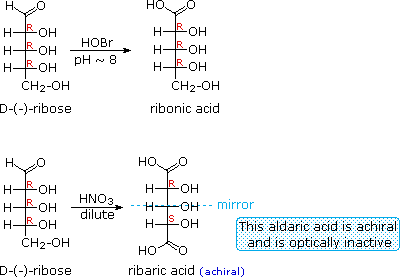
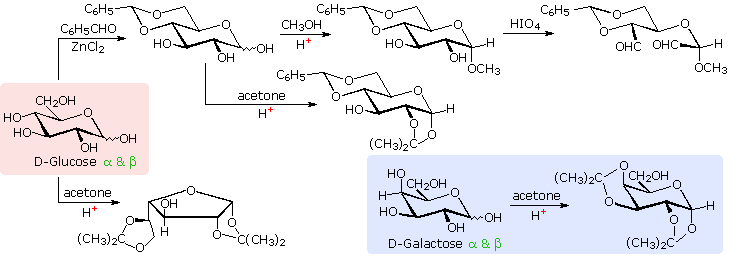
Il glucosio e altri saccaridi sono ampiamente scissi dall'acido periodico, grazie all'abbondanza di dioli vicinali nella loro struttura. Questa rimozione ossidativa, nota come reazione di Malaprade è particolarmente utile per l'analisi selettiva di derivati O-sostituiti dei saccaridi, dato che le funzioni eteree non reagiscono. La stechiometria della scissione di un aldoesoso è mostrata nella seguente equazione.
HOCH2(CHOH)4CHO + 5 HIO4 → H2C=O + 5 HCO2H + 5 HIO3
La configurazione del glucosio
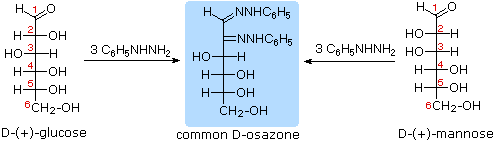
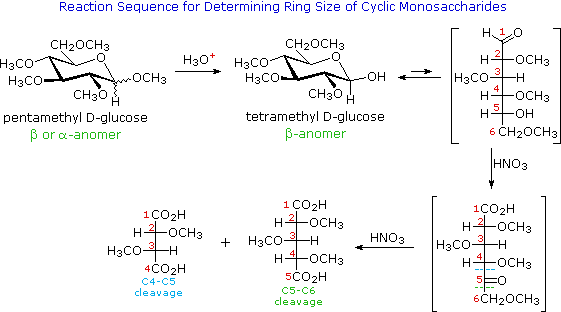
I quattro centri chirali nel glucosio indicato che possono esistere fino a sedici (24) stereoisomeri aventi questa costituzione. Questi dovrebbero esistere come otto coppie diastereoisomeriche di enantiomeri, e la sfida iniziale fu il determinare quale delle otto corrispondeva al glucosio. Questa sfida fu accettata e affrontata nel 1891 dal chimico tedesco Emil Fischer. Per lui fu una grande sfida, un po' come uscire da questo labirinto stereochimico, fu un tour de force logico, e alla fine gli valse nel 1902 il Premio Nobel per la chimica. Uno dei primi compiti affrontati da Fischer fu l'ideare un metodo per rappresentare la configrazione di ogni centro chirale in una maniera non ambigua. Alla fine, inventò una semplice tecnica per disegnare la catene di centri chirali, che ora noi chiamiamo la formulla di proiezione di Fischer. Ma di questo ne ho già parlato.
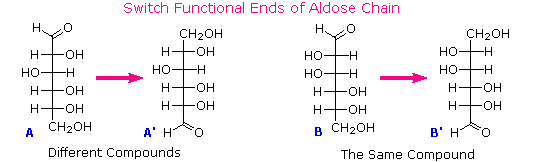
Al tempo in cui Fischer intraprese il progetto sul glucosio non era possibile stabilire la configurazione assoluta di un enantiomero. Di conseguenza, Fischer fece una scelta arbitraria per il (+)-glucosio e stabilì una rete di configurazioni di aldosi correlati che lui chiamò la famiglia D. Le immagini speculari di queste configurazioni furono quindi designate la famiglia L degli aldosi. Le formule delle proiezioni di Fischer ed i nomi per la famiglia dei D-aldosi (da tre a sei atomi di carbonio) sono mostrate sotto, con gli atomi di carbonio asimmetrici (centri chirali) colorati in rosso. L'ultimo centro chirale in una catena aldosa (il più lontano dal gruppo aldeidico) fu scelto da Fischer come sito designatore della famiglia D/L. Se il gruppo idrossile nella formula di proiezione punta verso destra, esso veniva definito come un membro della famiglia D. Un gruppo idrossile diretto verso sinistra (l'immagine speculare) rappresentava allora la famiglia L.
L'assegnazione iniziale di Fischer della configurazione D aveva un possibilità 50:50 di essere giusta, ma tutte le sue conclusioni successive riguardanti le configurazioni dei vari aldosi furono basate solidamente.
Nel 1951 studi di fluorescenza ai raggi X dell'acido (+)-tartarico, condotti in Olanda da Johannes Martin Bijvoet, provarono che la scelta di Fischer era corretta.
È importante riconoscere che il segno di una rotazione specifica di un composto (un numero sperimentale) non è correlato alla sua configurazione (D o L). È una questione semplice misurare una rotazione ottica con un polarimetro. La determinazione di una configurazione assoluta di solito richiede interconversioni chimiche con composti noti tramite cammini di reazione stereospecifici.
Sopra sono miostrati i prodotti proposti e la struttura generale del glucosio. I quattro atomi di carbonio centrali sono centri chirali e sono colorati in rosso.
Il glucosio e altri saccaridi sono ampiamente scissi dall'acido periodico, grazie all'abbondanza di dioli vicinali nella loro struttura. Questa rimozione ossidativa, nota come reazione di Malaprade è particolarmente utile per l'analisi selettiva di derivati O-sostituiti dei saccaridi, dato che le funzioni eteree non reagiscono. La stechiometria della scissione di un aldoesoso è mostrata nella seguente equazione.
HOCH2(CHOH)4CHO + 5 HIO4 → H2C=O + 5 HCO2H + 5 HIO3
La configurazione del glucosio
I quattro centri chirali nel glucosio indicato che possono esistere fino a sedici (24) stereoisomeri aventi questa costituzione. Questi dovrebbero esistere come otto coppie diastereoisomeriche di enantiomeri, e la sfida iniziale fu il determinare quale delle otto corrispondeva al glucosio. Questa sfida fu accettata e affrontata nel 1891 dal chimico tedesco Emil Fischer. Per lui fu una grande sfida, un po' come uscire da questo labirinto stereochimico, fu un tour de force logico, e alla fine gli valse nel 1902 il Premio Nobel per la chimica. Uno dei primi compiti affrontati da Fischer fu l'ideare un metodo per rappresentare la configrazione di ogni centro chirale in una maniera non ambigua. Alla fine, inventò una semplice tecnica per disegnare la catene di centri chirali, che ora noi chiamiamo la formulla di proiezione di Fischer. Ma di questo ne ho già parlato.
Al tempo in cui Fischer intraprese il progetto sul glucosio non era possibile stabilire la configurazione assoluta di un enantiomero. Di conseguenza, Fischer fece una scelta arbitraria per il (+)-glucosio e stabilì una rete di configurazioni di aldosi correlati che lui chiamò la famiglia D. Le immagini speculari di queste configurazioni furono quindi designate la famiglia L degli aldosi. Le formule delle proiezioni di Fischer ed i nomi per la famiglia dei D-aldosi (da tre a sei atomi di carbonio) sono mostrate sotto, con gli atomi di carbonio asimmetrici (centri chirali) colorati in rosso. L'ultimo centro chirale in una catena aldosa (il più lontano dal gruppo aldeidico) fu scelto da Fischer come sito designatore della famiglia D/L. Se il gruppo idrossile nella formula di proiezione punta verso destra, esso veniva definito come un membro della famiglia D. Un gruppo idrossile diretto verso sinistra (l'immagine speculare) rappresentava allora la famiglia L.
L'assegnazione iniziale di Fischer della configurazione D aveva un possibilità 50:50 di essere giusta, ma tutte le sue conclusioni successive riguardanti le configurazioni dei vari aldosi furono basate solidamente.
Nel 1951 studi di fluorescenza ai raggi X dell'acido (+)-tartarico, condotti in Olanda da Johannes Martin Bijvoet, provarono che la scelta di Fischer era corretta.
È importante riconoscere che il segno di una rotazione specifica di un composto (un numero sperimentale) non è correlato alla sua configurazione (D o L). È una questione semplice misurare una rotazione ottica con un polarimetro. La determinazione di una configurazione assoluta di solito richiede interconversioni chimiche con composti noti tramite cammini di reazione stereospecifici.
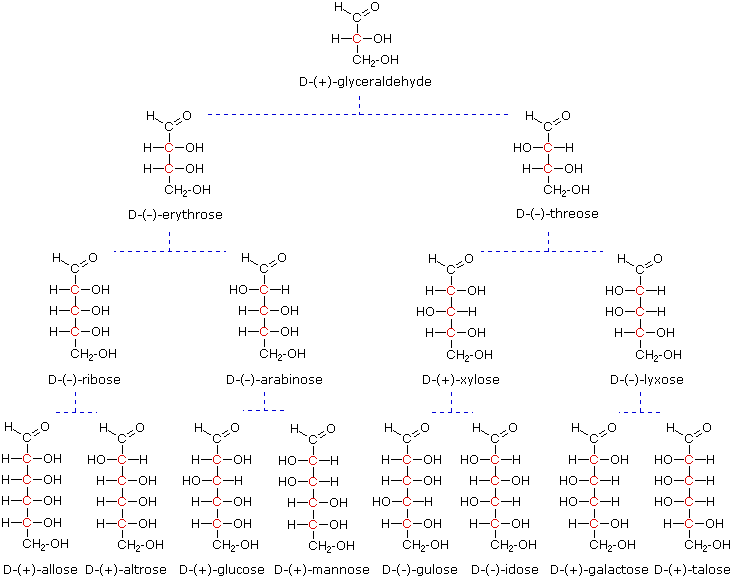
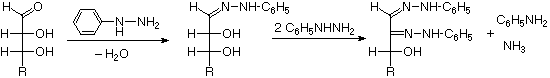
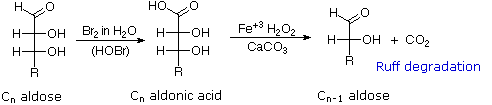
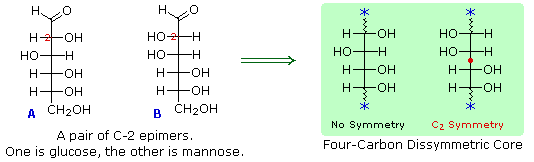
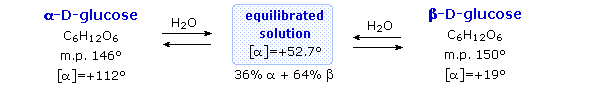
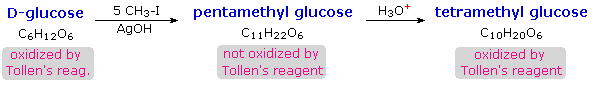
 L'amido è un polimero del glucosio, trovato in radici, rizomi, semi, steli, tuberi e bulbi delle piante, come granuli microscopici aventi caratteristiche forme e dimensioni. La maggior parte degli animali, inclusi gli umani, dipendono da questi amidi vegetali per il nutrimento. La struttura dell'amido è più complessa di quella della cellulosa. I granuli intatti sono insolubili in acqua fredda, ma triturandoli o mettendoli a bagno in acqua tiepida si causa la loro rottura.
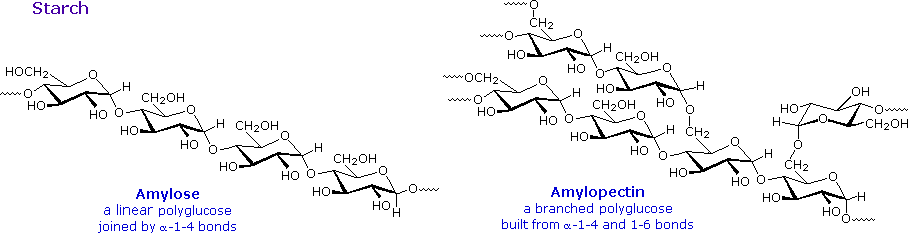
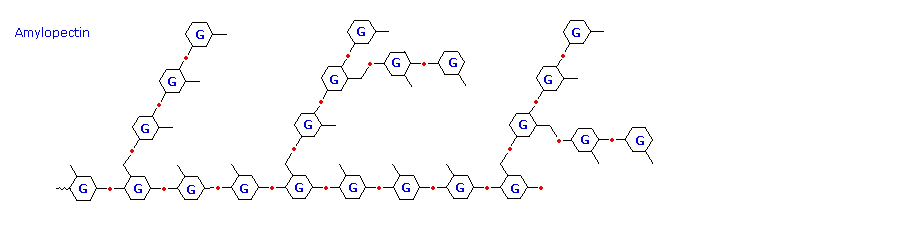
L'amido rilasciato consiste di due frazioni. Circa il 20% è un materiale solubile in acqua chiamato amilosio. Le molecule di amilossio sono catene lineari di diverse migliaia di unità di glucosio unite tramite legami α tra C-1 e C-4. Le soluzioni di amilosio sono attualmente dispersioni di micelle elicoidali idratate. La maggioranza dell'amido è una sostanza a peso molecolare molto più elevato, che consiste di circa un milione di unità di glucosio, ed è chiamata amilopectina. Le molecule di amilopectina sono reti ramificate costruite da connessioni glicosidiche da C-1 a C-4 e da C-1 a C-6, e sono essenzialmente insolubili in acqua.
Le formule strutturali rappresentative per amilosio e amilopectina sono mostrate sotto.
L'amido è un polimero del glucosio, trovato in radici, rizomi, semi, steli, tuberi e bulbi delle piante, come granuli microscopici aventi caratteristiche forme e dimensioni. La maggior parte degli animali, inclusi gli umani, dipendono da questi amidi vegetali per il nutrimento. La struttura dell'amido è più complessa di quella della cellulosa. I granuli intatti sono insolubili in acqua fredda, ma triturandoli o mettendoli a bagno in acqua tiepida si causa la loro rottura.
L'amido rilasciato consiste di due frazioni. Circa il 20% è un materiale solubile in acqua chiamato amilosio. Le molecule di amilossio sono catene lineari di diverse migliaia di unità di glucosio unite tramite legami α tra C-1 e C-4. Le soluzioni di amilosio sono attualmente dispersioni di micelle elicoidali idratate. La maggioranza dell'amido è una sostanza a peso molecolare molto più elevato, che consiste di circa un milione di unità di glucosio, ed è chiamata amilopectina. Le molecule di amilopectina sono reti ramificate costruite da connessioni glicosidiche da C-1 a C-4 e da C-1 a C-6, e sono essenzialmente insolubili in acqua.
Le formule strutturali rappresentative per amilosio e amilopectina sono mostrate sotto.
 La ramificazione in questo schema è esagerata, dato che in media, le ramificazioni avvengono solo ogni 25 unità di glucosio.
L'idrolisi dell'amido, di solito tramite reazioni enzimatiche, produce un liquido sciropposo che consiste largamente di glucosio. Quando l'amido di mais è la materia prima, questo prodotto è noto come sciroppo di mais. È largamente usato per ammorbidire tessuto, aggiungere volume, evitare cristallizzazione ed aumentare il sapore dei cibi.
Il glicogeno è il polimero che funge da deposito di glucosio, usato dagli animali. Ha una struttura simile all'amilopectina, ma anche molto più ramificato (circa ogni dieci unità di glucosio). Il grado di ramificazione in questi polisaccaridi può essere misurato tramite analisi enzimatica o chimica.
La ramificazione in questo schema è esagerata, dato che in media, le ramificazioni avvengono solo ogni 25 unità di glucosio.
L'idrolisi dell'amido, di solito tramite reazioni enzimatiche, produce un liquido sciropposo che consiste largamente di glucosio. Quando l'amido di mais è la materia prima, questo prodotto è noto come sciroppo di mais. È largamente usato per ammorbidire tessuto, aggiungere volume, evitare cristallizzazione ed aumentare il sapore dei cibi.
Il glicogeno è il polimero che funge da deposito di glucosio, usato dagli animali. Ha una struttura simile all'amilopectina, ma anche molto più ramificato (circa ogni dieci unità di glucosio). Il grado di ramificazione in questi polisaccaridi può essere misurato tramite analisi enzimatica o chimica.
 Modificazione sintetica della cellulosa
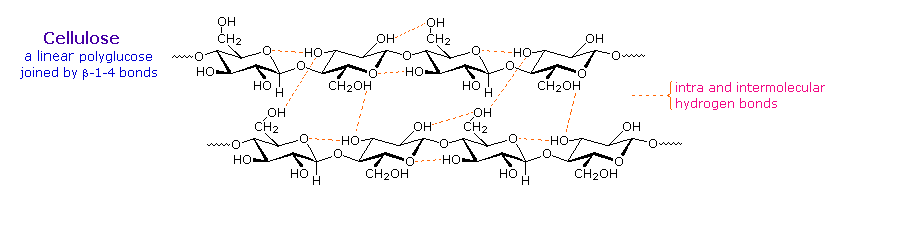
Il cotone, probabilmente la più utile fibra naturale, è cellulosa abbastanza pura. La produzione di tessuti dal cotone comporta la manipolazione fisica del materiale grezzo tramite cardatura, pettinatura e filatura di fibre selezionate. Per le stoffe il migliore cotone ha fibre lunghe, e fibre corte o polvere di cotone sono rimossi. La cellulosa grezza è anche disponibile dalla polpa del legno tramite dissoluzione della matrice lignano che la supporta. Queste fonte di cellulosa meno desiderate sono diffusamente usate per fare la carta.
Al fine di espandere i modi in cui la cellulosa può essere resa di uso pratico, i chimici hanno creato tecniche per la preparazione di soluzioni di derivati della cellulosa che possono essere filate in fibre, distese in un film o fuse in diverse forme solide. Un fattore chiave in queste trasformazioni sono i tre gruppi idrossile liberi su ogni unità di glucosio nella catena della cellulosa, —[C6H7O(OH)3]n—. L'esterificazione di queste funzioni porta a prodotti polimerici aventi proprietà davvero differenti comparate a quelle della cellulosa stessa.
Il nitrato di cellulosa, preparato per la prima volta più di 150 anni fa trattando la cellulosa con acido nitrico, è il primissimo polimero sintetico a vedere un uso generale. Il composto completamente nitrato, —[C6H7O(ONO2)3]n—, chiamato fulmicotone o cotone fulminante, è esplosivo ed infiammabile ed è un componente della polvere senza fumo. La cellulosa parzialmente nitrata è chiamata pirossilina. La pirossilina è solubile in etere e ai tempi era usata per pellicole di film/fotografiche e lacche. L'elevata infiammabilità della pirossilina causò molti incendi tragici nei cinema duranta il periodo del suo impiego. Inoltre, la lenta idrolisi della pirossilina porta all'acido nitrico, un processo che ha contribuito al deterioramento dei primi film durante l'immagazzinamento.
L'acetato di cellulosa, —[C6H7O(OAc)3]n—, è meno infiammabile della pirossilina, e l'ha sostituita nella maggior parte delle applicazioni. È preparato tramite reazione della cellulosa con anidride acetica ed un catalizzatore acido. Le proprietà del prodotto variano con il grado di acetilazione. Alcuni accorciamenti di catena avvendono inevitabilmente nelle preparazioni. Una soluzione in acetone di acetato di cellulosa può essere forzata attraverso un estrusore per generare filamenti: si ottiene così il rayon acetato, che può essere intrecciato in tessuti.
Il rayon viscoso, viene preparato tramite formazione di un derivato solubile xantato alcalino che può essere filato in una fibra che riforma cellulosa per quenching acido. La seguente equazione generale illustra queste trasformazioni. La fibra prodotta è chiamata rayon viscoso.
ROH + NaOH
Modificazione sintetica della cellulosa
Il cotone, probabilmente la più utile fibra naturale, è cellulosa abbastanza pura. La produzione di tessuti dal cotone comporta la manipolazione fisica del materiale grezzo tramite cardatura, pettinatura e filatura di fibre selezionate. Per le stoffe il migliore cotone ha fibre lunghe, e fibre corte o polvere di cotone sono rimossi. La cellulosa grezza è anche disponibile dalla polpa del legno tramite dissoluzione della matrice lignano che la supporta. Queste fonte di cellulosa meno desiderate sono diffusamente usate per fare la carta.
Al fine di espandere i modi in cui la cellulosa può essere resa di uso pratico, i chimici hanno creato tecniche per la preparazione di soluzioni di derivati della cellulosa che possono essere filate in fibre, distese in un film o fuse in diverse forme solide. Un fattore chiave in queste trasformazioni sono i tre gruppi idrossile liberi su ogni unità di glucosio nella catena della cellulosa, —[C6H7O(OH)3]n—. L'esterificazione di queste funzioni porta a prodotti polimerici aventi proprietà davvero differenti comparate a quelle della cellulosa stessa.
Il nitrato di cellulosa, preparato per la prima volta più di 150 anni fa trattando la cellulosa con acido nitrico, è il primissimo polimero sintetico a vedere un uso generale. Il composto completamente nitrato, —[C6H7O(ONO2)3]n—, chiamato fulmicotone o cotone fulminante, è esplosivo ed infiammabile ed è un componente della polvere senza fumo. La cellulosa parzialmente nitrata è chiamata pirossilina. La pirossilina è solubile in etere e ai tempi era usata per pellicole di film/fotografiche e lacche. L'elevata infiammabilità della pirossilina causò molti incendi tragici nei cinema duranta il periodo del suo impiego. Inoltre, la lenta idrolisi della pirossilina porta all'acido nitrico, un processo che ha contribuito al deterioramento dei primi film durante l'immagazzinamento.
L'acetato di cellulosa, —[C6H7O(OAc)3]n—, è meno infiammabile della pirossilina, e l'ha sostituita nella maggior parte delle applicazioni. È preparato tramite reazione della cellulosa con anidride acetica ed un catalizzatore acido. Le proprietà del prodotto variano con il grado di acetilazione. Alcuni accorciamenti di catena avvendono inevitabilmente nelle preparazioni. Una soluzione in acetone di acetato di cellulosa può essere forzata attraverso un estrusore per generare filamenti: si ottiene così il rayon acetato, che può essere intrecciato in tessuti.
Il rayon viscoso, viene preparato tramite formazione di un derivato solubile xantato alcalino che può essere filato in una fibra che riforma cellulosa per quenching acido. La seguente equazione generale illustra queste trasformazioni. La fibra prodotta è chiamata rayon viscoso.
ROH + NaOH  RO‒ Na+ + S=C=S
RO‒ Na+ + S=C=S  RO-CS2‒Na+ + H3O+
RO-CS2‒Na+ + H3O+ 
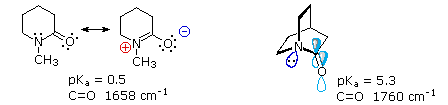
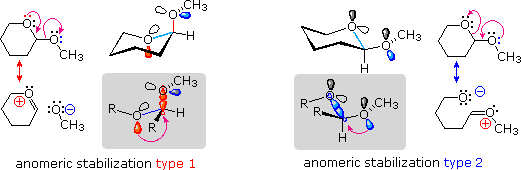
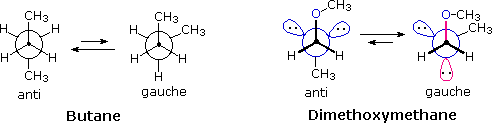






 e serviva al gruppo di ricerca in cui ero quando lavoravo sui glicosidi.
e serviva al gruppo di ricerca in cui ero quando lavoravo sui glicosidi.
 oppure ti fanno storie (giuste o meno) se compri più grammi di taluni reagenti come la 3,4-(metilenediossi)benzaldeide
oppure ti fanno storie (giuste o meno) se compri più grammi di taluni reagenti come la 3,4-(metilenediossi)benzaldeide 
{kind=link}
{kind=link}
{kind=link}