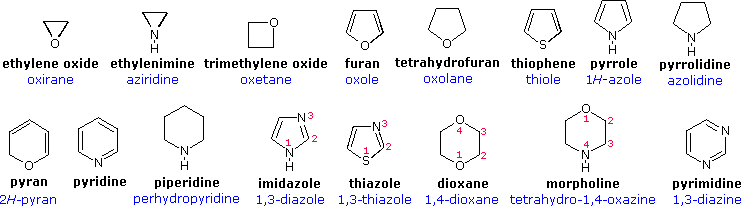
Anelli a cinque membri
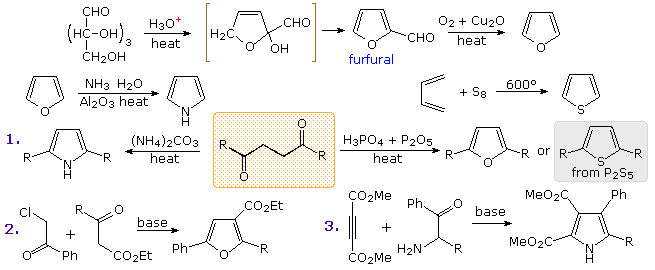
La preparazione commerciale del furano avviene passando attraverso l'aldeide, furfurale, che a sua volta è generata da materiali grezzi contenenti pentosio come gli scarti della produzione del mais, come mostrato nell'equazione più in alto dello schema sotto. Preparazioni simili del pirrolo e del tiofene sono illustrate nella seconda fila di equazioni.
L'equazione 1 nella terza fila illustra una preparazione generale di furani, pirroli e tiofeni sostituiti, da composti 1,4-dicarbonilici, nota come sintesi di Paal-Knorr.
Molte altre procedure che portano ad eterocicli sostituiti di questo tipo sono state ideate. Due di queste sono mostrate nelle reazioni 2 e 3.
Il furano è ridotto a tetraidrofurano tramite idrogenazione palladio-catalizzata. Questo etere ciclico non è solo un solvente valido, ma è anche prontamente convertito a 1,4-dialogenobutani o 4-alogenoalchilsolfonati, che possono essere usati per preparare pirrolidina e tiolano.
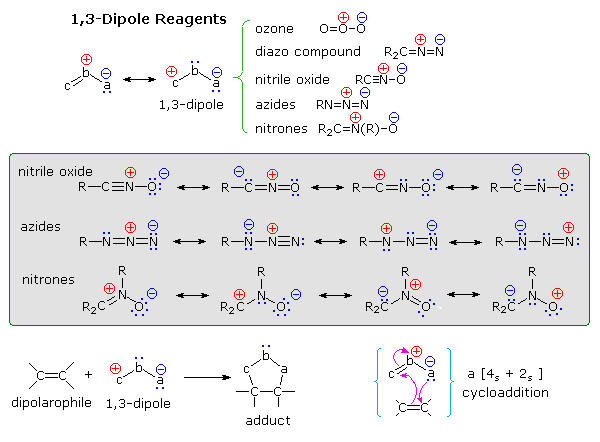
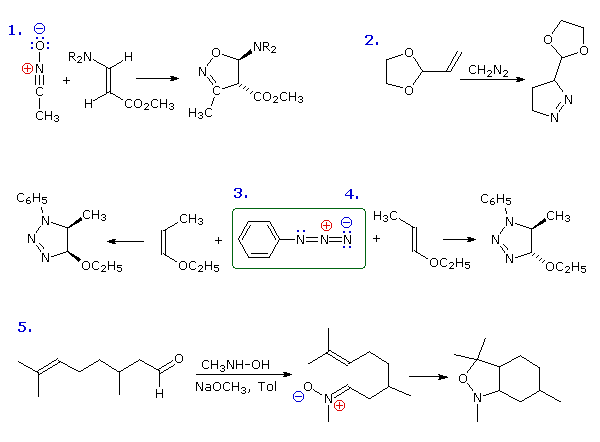
Le reazioni di cicloaddizione dipolare spesso portano ad eterocicli a cinque termini più complessi. Ma di questo ne parlerò dopo.
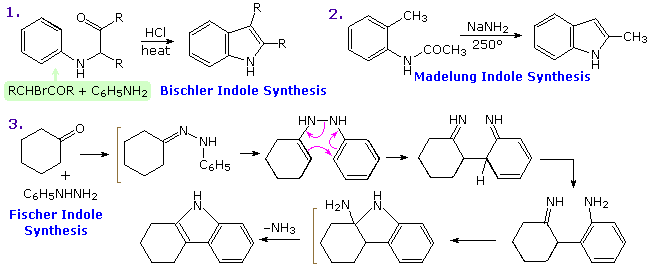
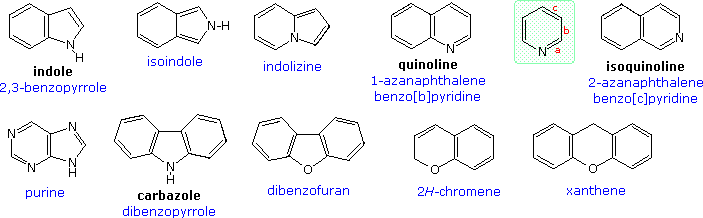
L'indolo è probabilmente l'anello eterociclico fuso più importante in questa classe. Nello schema sotto sono riportati tre esempi di sintesi indolica.
Il primo procede attraverso sostituzione elettrofila di un anello benzenico attivato da azoto.
Il secondo presumibilmente avviene tramite formazione di una specie dianionica in cui l'unità ArCH2– si lega al gruppo carbonile disattivato.
Infine, la sintesi indolica di Fischer è una sequenza degna di nota di reazioni: tautomerismo, riarrangiamento sigmatropico, addizione nucleofila, ed eliminazione che avvengono in seguito alla formazione del fenilidrazone. Questa interessante trasformazione coinvolge l'ossidazione di due atomi di carbonio e la riduzione di un carbonio e di entrambi gli atomi di azoto.
La reattività chimica dei membri saturi di questa classe di eterocicli: tetraidrofurano, tiolane e pirrolidina, assomiglia a quella degli etere, solfuri, ed amine secondarie acicliche, ma non verrà descritta qui.
Gli 1,3-diossolani ed i ditiolani sono acetali e tioacetali ciclici. Queste unità sono comunemente usate come gruppi protettivi per aldeidi e chetoni, e possono essere idrolizzate tramite azione di acidi acquosi.
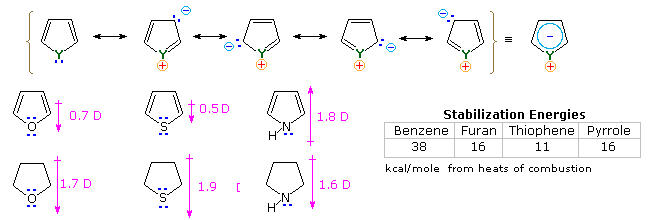
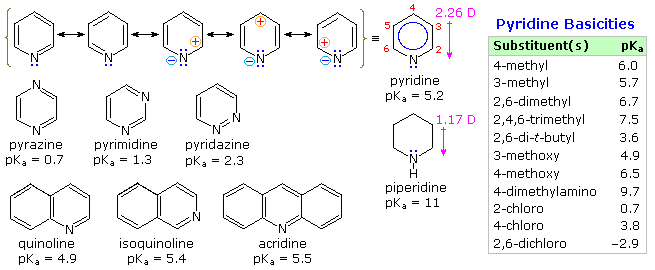
Sono i composti insaturi "aromatici" furano, tiofene e pirrolo che richiedono la nostra attenzione. In ogni caso l'eteroatomo ha almeno una coppia di elettroni non leganti che può combinarsi con i quattro elettroni π dei doppi legami per produrre un annulene avente un sestetto aromatico di elettroni. Questo è illustrato tramite la descrizione di risonanza nella parte alta del seguente schema. L'eteroatomo Y diventa ibridizzato sp2 ed acquista una carica positiva dato che la sua coppia elettronica è delocalizzata attorno all'anello. Una conseguenza facilmente osservata di questa delocalizzazione è un cambiamento nel momento dipolare comparato con gli analoghi eterocicli saturi, che hanno tutti forti dipoli con l'eteroatomo all'estremità negativa. Come previsto, gli eterociclici aromatici hanno momenti dipolari molto più piccoli, o nel caso del pirrolo un grande dipolo nella direzioni opposta.
Un'importante caratteristica dell'aromaticità è l'aumentata stabilità termodinamica, e questo è di solito dimostrato dalle misure dei relativi calori di idrogenazione o dei calori di combustione. Tramite questo standard, i tre eterocicli aromatici sotto esame sono stabilizzati, ma con un minore grado rispetto al benzene.
Ulteriori prove del carattere aromatico del pirrolo è riscontrato nella sua eccezionalmente debole basicità (pKa ca. 0) e forte acidità (pKa = 15) per un'amina secondaria. I corrispondenti valori per l'amina satura pirrolidina sono: basicità 11.2 ed acidità 32.
Un'altra caratteristica dei sistemi aromatici, di particolare importanza per i chimici, è il loro modello di reattività con reagenti elettrofili. Mentre i semplici cicloalcheni generalmente danno reazioni di addizione, i composti aromatici tendono reagire tramite sostituzione. Come notato per il benzene ed i suoi derivati, queste sostituzioni avvengono tramite un'iniziale addizione elettrofila, seguita da una perdita di protone dall'intermedio "onio" per rigenerare l'anello aromatico.
Gli eterocicli aromatici a cinque membri subiscono tutti sostituzione elettrofila, con un ordine generale di reattività: pirrolo >> furano > tiofene > benzene.
Alcuni esempi sono dati nel seguente schema.
Le condizioni di reazione mostrano chiaramente la maggiore reattività del furano comparata col tiofene. Tutti questi eterocicli aromatici reagiscono vigorosamente con cloro e bromo, spesso formando prodotti polialogenati assieme a polimeri. L'eccezionale reattività del pirrolo è evidenziata dalla sua reazione con lo iodio (equazione in basso a sinistra), e la formazione del 2-acetilpirrolo per semplice riscaldamento con anidride acetica (senza catalizzatore).
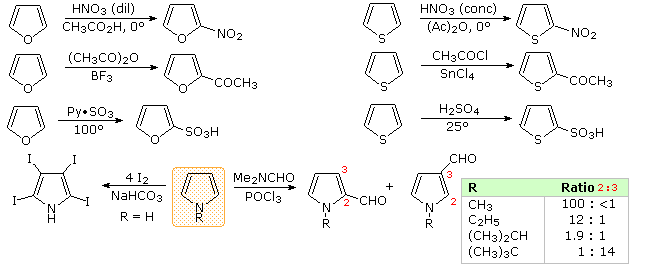
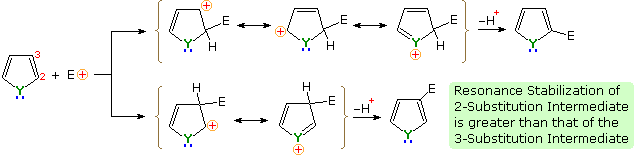
C'è una chiara preferenza per la sostituzione in posizione 2 (α) dell'anello, specialmente per furano e tiofene. Le reazioni del pirrolo richiedono una attenta valutazione, dato che la N-protonazione distrugge il suo carattere aromatico. Inoltre, l'N-sostituzione di questa amina secondaria è spesso condotta prima delle reazioni successive. Per esempio, il pirrolo reagisce con anidride acetica o acetile cloruro e trietilamina a dare l'N-acetilpirrolo. Di conseguenza, la regioselettività della sostituzione sul pirrolo è variabile, come notato dall'equazione in basso a destra.
Una spiegazione per la selettività α generale di queste sostituzione è chiaro dal meccanismo sottolineato sotto. L'intermedio formato dall'attacco nucleofilo al C-2 è stabilizzato tramite delocalizzazione di carica con un maggiore grado rispetto all'intermedio derivato dall'attacco al C-3 attack. Dal postulato di Hammond possiamo quindi desumere che l'energia di attivazione per la sostituzione alla posizione prima è minore di quella per la posizione ultima.
I sostituenti funzionali influenzano le reazioni di sostituzione di questi eterocicli in una maniera molto simile a quello che succede per il benzene. Inoltre, non appena uno capisce il carattere orto-para e meta orientante di questi sostituenti, la loro influenza direzionale sulla sostituzione sull'anello eterociclico non è difficile da predire.
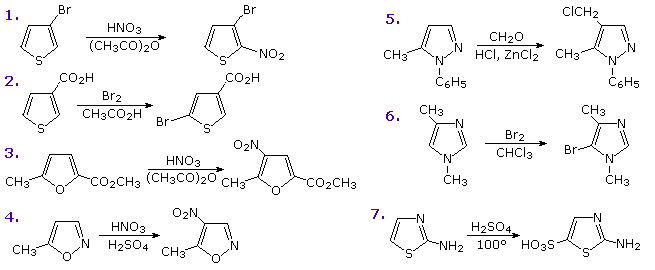
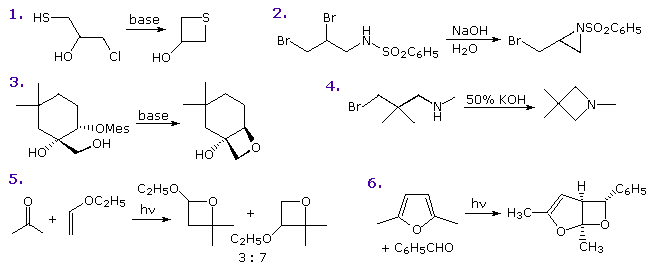
Il seguente schema mostra sette di tali reazioni.
Le reazioni 1 & 2 sono tiofeni sostituiti in posizione 3, il primo da un sostituente elettron donatore ed il secondo da un gruppo elettron attrattore.
La terza reazione ha due sostituenti di diverso tipo in posizione 2 e 5.
Infine, gli esempi da 4 a 7 illustrano reazioni dell'1,2- e dell'1,3-ossazolo, tiazolo e diazolo. Notate che la basicità dell'azoto ibridizzato
sp2 nei diazoli è più di un milione di volte più grande dell'azoto ibridizzato apparentemente sp
3, la coppia elettronica del quale è parte del sestesso aromatico di elettroni.
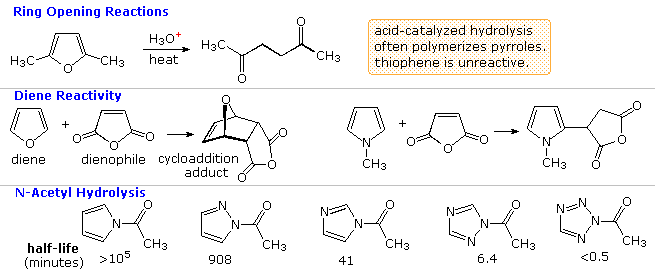
Altre possibili reazioni sono suggerite dalle caratteristiche strutturali di questi eterocicli. Per esempio, il furano potrebbe essere considerato un enol etere ed il pirrolo una enamina. Tali funzioni sono note subire idrolisi acido catalizzata di composti carbonilici e ad alcoli o amine. Dato che questi composti sono anche dieni sostituiti con eteroatomi, potremmo anticipare le reazioni di cicloaddizione di Diels-Alder con dienofili appropriati. Queste possibilità saranno illustrate sotto. Come notato nell'esempiopiù sopra, i furani possono inoltre essere idrolizzati a composti 1,4-dicarbonilici, ma pirroli e tiofeni si comporta in modo differente.
I secondi due esempi, mostrati nel mezzo, dimostrano le tipiche reazioni di furano e pirrolo con il forte dienofilo anidride maleica. Il primo participa in una reazione di cicloaddizione; comunque, il pirrolo subisce semplicemente sostituzione elettrofila al C-2. Il tiofene non reagisce facilmente con questo dienofilo.
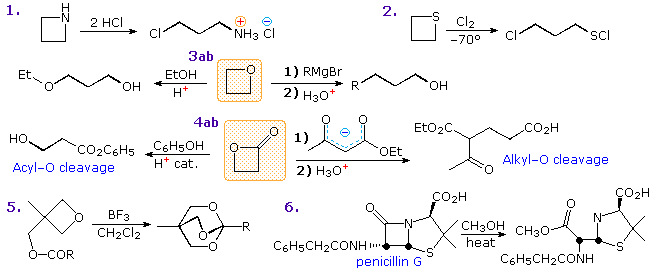
La linea inferiore di questo schema illustra la notevole influenza che le unità di azoto addizionali hanno sull'idrolisi di una serie di
N-acetilazoli in acqua a 25 ºC e pH=7. Il composto del pirrolo sulla sinistra è essenzialmente non reattivo, come previsto per un'ammide, ma gli azoti addizionali aumenta la velocità dell'idrolisi. Questo effetto è stato reso di uso pratico nell'applicazione del reagente acilante 1,1'-carbonildiimidazolo (reagente di Staab).
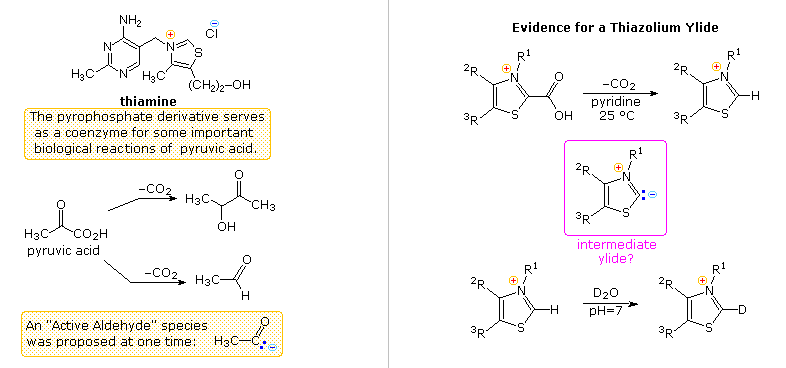
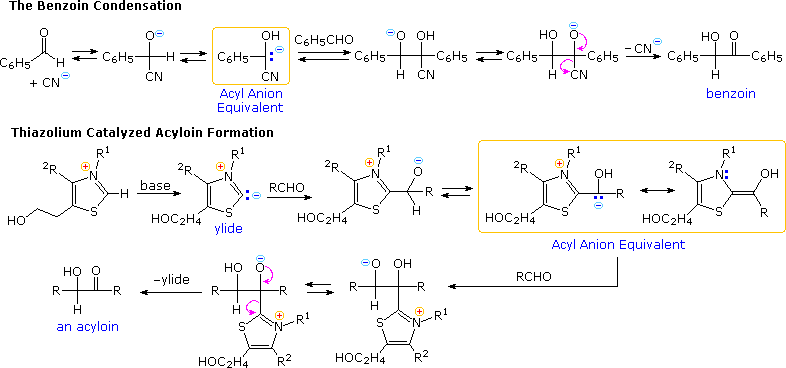
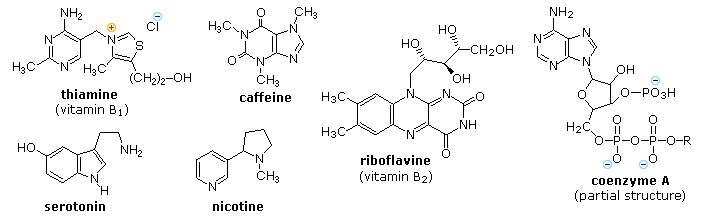
Un altro aspetto della chimica eterociclica è stato svelato nel corso delle indagini riguardanti l'azione della tiamina (schema seguente). Come il suo derivato pirofosfato, la tiaminaa è un coenzima per diverse reazioni biochimiche, particolarmente decarbossilazioni dell'acido piruvico ad acetaldeide ed acetoina. I primi lavoratori supponeva che una "aldeide attivata" o una specie acil carbanionica fosse un intermedio in queste reazioni. Furono fatte molte proposte, alcune che coinvolgevano la metà aminopirimidinica, ed altre, derivati di idrolisi apertura d'anello dell'anello tiazolico, ma nessuna era soddisfacente.
Questo enigma fu risolto da R. Breslow (Columbia) che trovò che l'idrogeno in C-2 dei sali di tiazolio era inaspettatamente acido (pK
a ca. 13), formando una base coniugata relativamente stabile, un'ilide. Come mostrato, questo razionalizza la semplice decarbossilazione degli acidi tiazolio-2-carbossilici e lo scambio di deuterio al C-2 in acqua pesante neutra.
Gli appropriati sali di tiazolio catalizzano la conversione di aldeidi in aciloine più o meno lo stesso modo in cui lo ione cianuro catalizza la formazione del benzoino dalla benzaldeide, la condensazione benzoinica.
Sotto è mostrato il meccanismo per queste due reazioni. Notate che in entrambi i casi un equivalente di anione acile è formato e quindi si addiziona alla funzione carbonilica nella maniera prevista.
La condensazione benzoinica è limitata alle aldeidi aromatiche, ma l'uso di catalizzatori tiazolio si è dimostrato in generale efficace per aldeidi alifatiche ed aromatiche. Questo approccio alle aciloine impiega condizioni più blande rispetto alla riduzione degli esteri ad intermedi enediolici tramite l'azione di sodio metallico.
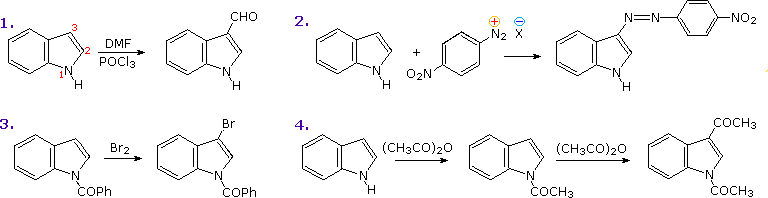
Il più importante sistema ciclico condensato correlato a questi eterocicli è l'indolo. Alcune reazioni di sostituzione elettrofila dell'indolo sono mostrate nello schema sotto. Se l'azoto dell'indolo è sostituito o meno, il sito favorito per l'attacco è il C-3 dell'anello eterociclico. Il legame di questo elettrofilo a quella posizione permette la stabilizzazione dell'intermedio onio tramite l'azoto senza rottura dell'aromaticità del benzene.

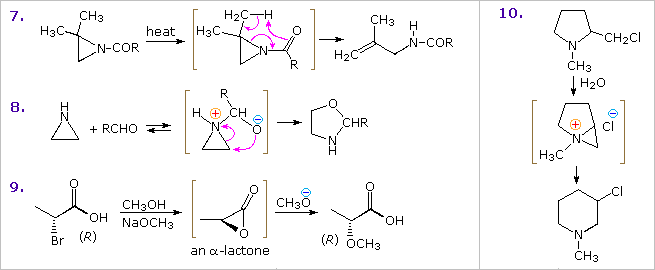
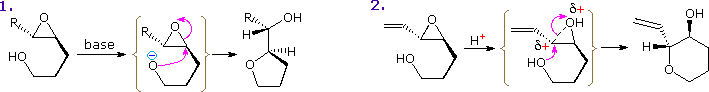
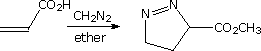
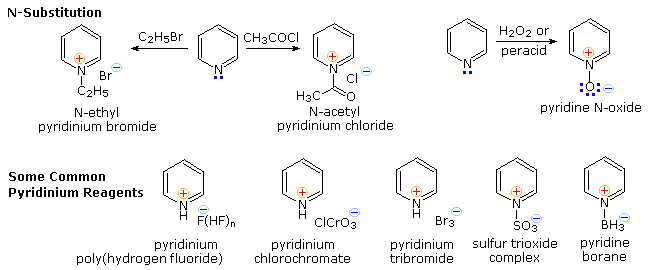
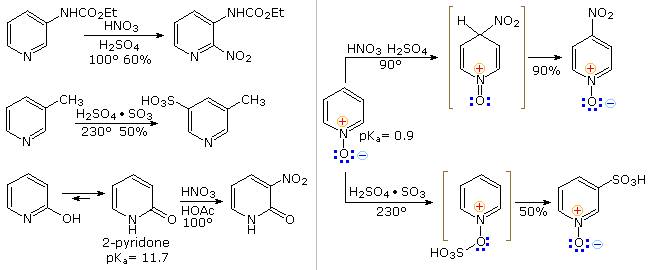
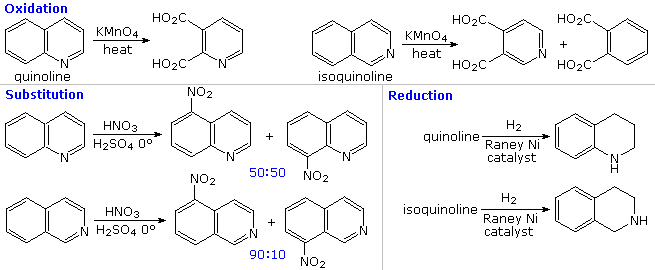
 Sotto sono mostrati altri cinque esempi di reazioni di una base o un nucleofilo con piridine sostituite. Poiché l'anello della piridina (e ancor meglio l'anello N-ossido) possono mantenere una carica negativa, i sostituenti alchilici in posizione 2 e 4 sono attivati allo stesso modo di un gruppo carbonile.
Le reazioni 6 e 7 mostrano l'alchilazione e reazioni di condensazione risultanti da questa attivazione.
La reazione 8 è un esempi di formazione di N-alchilpiridone tramite addizione di idrossido ad un catione N-alchil piridinio, seguita da leggera ossidazione.
La riduzione di Birch converte le piridine in diidropiridine che sono bis-enamine e possono essere idrolizzata a composti 1,5-dicarbonilici. I sali di piridinio subiscono un SET (single electron transfer) generando radicali liberi notevolmente stabili.
L'esempio mostrato nella reazione 9 è un liquido verde distillabile, stabile (in assenza di ossigeno).
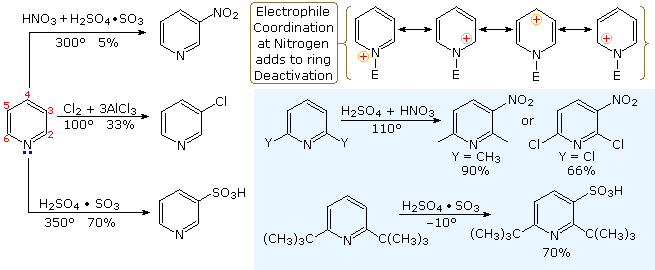
Sebbene le 3-alogenopiridine non subiscano reazioni di sostituzione del tipo addizione-eliminazione come fanno gli isomeri 2 e 4, la base forte sodio ammide effettua aminazione passando da un intermedio piridino. Questo è illustrato dalla reazione 10. È interessante che il 3-piridino sia formato in presenza di 2-piridino. L'ultimo è formato se il C-4 è occupat da un sostituente alchilico.
L'intermedio piridino è simile al benzino. Ed è un esempio di sostituzione del tipo eliminazione-addizione, analoga a quella che avviene col benzino.
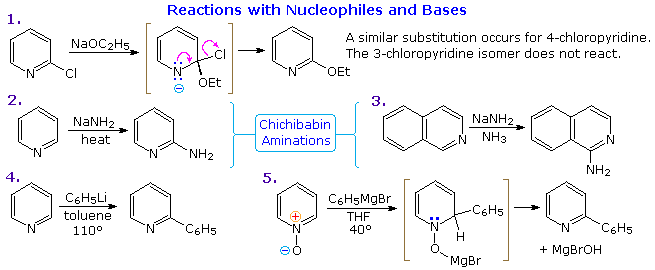
Sotto sono mostrati altri cinque esempi di reazioni di una base o un nucleofilo con piridine sostituite. Poiché l'anello della piridina (e ancor meglio l'anello N-ossido) possono mantenere una carica negativa, i sostituenti alchilici in posizione 2 e 4 sono attivati allo stesso modo di un gruppo carbonile.
Le reazioni 6 e 7 mostrano l'alchilazione e reazioni di condensazione risultanti da questa attivazione.
La reazione 8 è un esempi di formazione di N-alchilpiridone tramite addizione di idrossido ad un catione N-alchil piridinio, seguita da leggera ossidazione.
La riduzione di Birch converte le piridine in diidropiridine che sono bis-enamine e possono essere idrolizzata a composti 1,5-dicarbonilici. I sali di piridinio subiscono un SET (single electron transfer) generando radicali liberi notevolmente stabili.
L'esempio mostrato nella reazione 9 è un liquido verde distillabile, stabile (in assenza di ossigeno).
Sebbene le 3-alogenopiridine non subiscano reazioni di sostituzione del tipo addizione-eliminazione come fanno gli isomeri 2 e 4, la base forte sodio ammide effettua aminazione passando da un intermedio piridino. Questo è illustrato dalla reazione 10. È interessante che il 3-piridino sia formato in presenza di 2-piridino. L'ultimo è formato se il C-4 è occupat da un sostituente alchilico.
L'intermedio piridino è simile al benzino. Ed è un esempio di sostituzione del tipo eliminazione-addizione, analoga a quella che avviene col benzino.
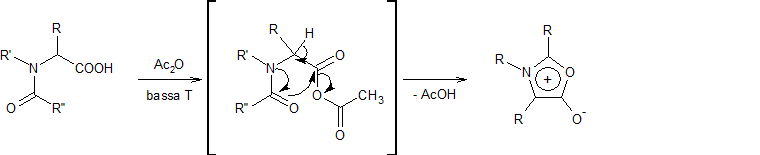




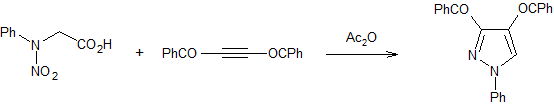
 della peptidomimetica*, mi sono imbattuto in una delle molte, ma non troppe
della peptidomimetica*, mi sono imbattuto in una delle molte, ma non troppe 











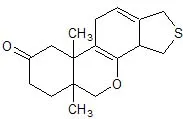
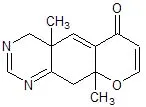
 Ma non ho problemi a ritornare sull'argomento... Devo solo trovare dove è, e vedere se va bene... Se no devo cercare una fonte autorevole. Non è cosa semplice come si è visto numerare eterocicli complessi
Ma non ho problemi a ritornare sull'argomento... Devo solo trovare dove è, e vedere se va bene... Se no devo cercare una fonte autorevole. Non è cosa semplice come si è visto numerare eterocicli complessi 