1. Nucleofilicità dei composti dello zolfo
Gli analoghi allo zolfo degli alcoli sono chiamati tioli o mercaptani, e gli analoghi degli eteri sono chiamati solfuri. Il comportamento chimico di tioli e solfuri è in contrasto con quello di alcoli ed eteri in alcuni importanti modi. Dato che il solfuro di idrogeno (H2S) è un acido molto più forte dell'acqua (più di dieci milioni di volte), ci si aspetta, e si trova poi, che il tioli sono acidi più forti degli equivalenti alcoli e fenoli. Le basi coniugate tiolato vengono formate facilmente, e si sono dimostrate essere nucleofili eccellenti in reazioni SN2 di alogenuri alchilici e tosilati.
R–S–Na+ + (CH3)2CH–Br → (CH3)2CH–S–R + Na+Br–
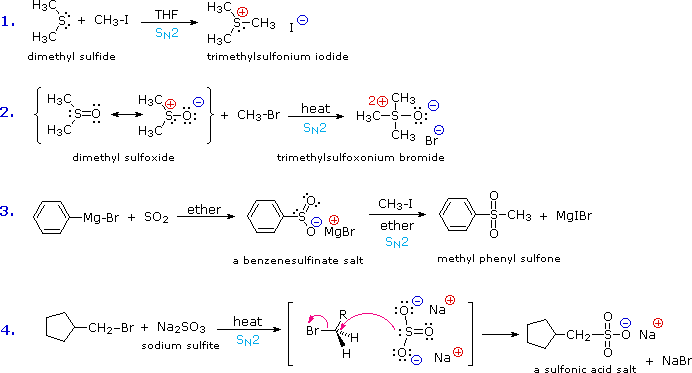
Sebbene la basicità degli eteri sia approssimativamente un centinaio di volte più grande di quella degli equivalenti solfuri, la nucleofilicità dello zolfo è molto più grande di quella dell'ossigeno; quindi esiste un numero di interessanti ed utili sostituzioni elettrofile dello zolfo che non è normalmente osservato per l'ossigeno. I solfuri, per esempio, reagiscono con alogenuri alchilici a dare sali di solfonio ternari (equazione #1) nello stesso modo in cui le amine terziarie vengono alchilate a sali di ammonio quaternari. Sebbene siano noti gli equivalenti sali di ossonio degli eteri, essi vengono preparati solo in condizioni estreme, e sono eccezionalmente reattivi. Incredibilmente, anche i solfossidi (equazione #2), i sali sulfinato (#3) e gli anioni solfito (#4) alchilano con lo zolfo, nonostante la parziale carica negativa formale sull'ossigeno e la parziale carica positiva sullo zolfo.
 2. Stati di ossidazione dei composti dello zolfo
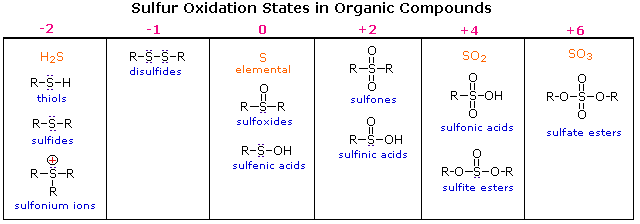
L'ossigeno assume solo due stati di ossidazione nei suoi composti organici (–1 in perossidi e –2 negli altri composti). Zolfo, dall'altra parte, si trova negli stati di ossidazione che vanno da –2 a +6, come mostrato nella tabella seguente (alcuni composti organici semplici sono mostrati in arancio).
2. Stati di ossidazione dei composti dello zolfo
L'ossigeno assume solo due stati di ossidazione nei suoi composti organici (–1 in perossidi e –2 negli altri composti). Zolfo, dall'altra parte, si trova negli stati di ossidazione che vanno da –2 a +6, come mostrato nella tabella seguente (alcuni composti organici semplici sono mostrati in arancio).
 Restringendo le formule agli elettroni del guscio di valenza, la maggior parte degli stati di ossidazione più elevati avranno una separazione di carica, come nell'equazione 2 sopra. Le formule scritte qui neutralizzano questa separazione di carica tramite doppi legami che espandono l'ottetto di valenza dello zolfo. Inoltre, i doppi legami S=O non consistono degli abituali orbitali σ & π che si trovano nei doppi legami carbonio-carbonio. Essendo un elemento del terzo periodo, lo zolfo ha cinque orbitali 3d vuoti che possono essere usati per il legame p-d in modo simile al legame p-p (π). In questo modo lo zolfo può espandere l'ottetto di valenza tipo-argo di due (solfossidi) o quattro (solfoni) elettroni. I solfossidi hanno una forma piramidale rigida (il doppietto non legante dello zolfo occupa un angolo di un tetraedro con lo zolfo al centro). Di conseguenza, i solfossidi aventi due diversi sostituenti alchilici o arilici sono chirali. I solfossidi enantiomerici sono stabili e possono essere isolati.
Anche i tioli differiscono drammaticamente dagli alcoli nella loro chimica dell'ossidazione. L'ossidazione di alcoli primari e secondari ad aldeidi e chetoni cambia lo stato di ossidazione del carbonio ma non dell'ossigeno. L'ossidazione dei tioli e degli altri composti dello zolfo cambia lo stato di ossidazione dello zolfo piuttosto che quello del carbonio. Negli esempi che seguono sono rappresentate ossidazioni dello zolfo. Nel primo caso, un'ossidazione blanda converte i tioli a disolfuri. Un'ossidazione di questo tipo degli alcoli a perossidi non è normalmente osservata. Le ragioni di questo comportamento differente non sono difficili da intuire. Il legame singolo S–S è circa due volte più forte del legame O–O nei perossidi, e il legame O–H è più forte di 25 kcal/mole di un legame S–H. Quindi, la termodinamica favorisce la formazione dei disolfuri rispetto ai perossidi.
La blanda ossidazione dei disolfuri con cloro porta ad alchilsulfenil cloruri, ma un'ossidazione più energica forma gli acidi solfonici (2° esempio). Infine, l'ossidazione dei solfuri con perossido di idrogeno (o peracidi) prima porta ai solfossidi e poi ai solfoni; se si vuole passare direttamente da solfuro a solfone si può impiegare il permanganato di potassio. Un'altra interessante ossidazione di solfuri a solfossidi si può effettuare con sodio periodato.
Restringendo le formule agli elettroni del guscio di valenza, la maggior parte degli stati di ossidazione più elevati avranno una separazione di carica, come nell'equazione 2 sopra. Le formule scritte qui neutralizzano questa separazione di carica tramite doppi legami che espandono l'ottetto di valenza dello zolfo. Inoltre, i doppi legami S=O non consistono degli abituali orbitali σ & π che si trovano nei doppi legami carbonio-carbonio. Essendo un elemento del terzo periodo, lo zolfo ha cinque orbitali 3d vuoti che possono essere usati per il legame p-d in modo simile al legame p-p (π). In questo modo lo zolfo può espandere l'ottetto di valenza tipo-argo di due (solfossidi) o quattro (solfoni) elettroni. I solfossidi hanno una forma piramidale rigida (il doppietto non legante dello zolfo occupa un angolo di un tetraedro con lo zolfo al centro). Di conseguenza, i solfossidi aventi due diversi sostituenti alchilici o arilici sono chirali. I solfossidi enantiomerici sono stabili e possono essere isolati.
Anche i tioli differiscono drammaticamente dagli alcoli nella loro chimica dell'ossidazione. L'ossidazione di alcoli primari e secondari ad aldeidi e chetoni cambia lo stato di ossidazione del carbonio ma non dell'ossigeno. L'ossidazione dei tioli e degli altri composti dello zolfo cambia lo stato di ossidazione dello zolfo piuttosto che quello del carbonio. Negli esempi che seguono sono rappresentate ossidazioni dello zolfo. Nel primo caso, un'ossidazione blanda converte i tioli a disolfuri. Un'ossidazione di questo tipo degli alcoli a perossidi non è normalmente osservata. Le ragioni di questo comportamento differente non sono difficili da intuire. Il legame singolo S–S è circa due volte più forte del legame O–O nei perossidi, e il legame O–H è più forte di 25 kcal/mole di un legame S–H. Quindi, la termodinamica favorisce la formazione dei disolfuri rispetto ai perossidi.
La blanda ossidazione dei disolfuri con cloro porta ad alchilsulfenil cloruri, ma un'ossidazione più energica forma gli acidi solfonici (2° esempio). Infine, l'ossidazione dei solfuri con perossido di idrogeno (o peracidi) prima porta ai solfossidi e poi ai solfoni; se si vuole passare direttamente da solfuro a solfone si può impiegare il permanganato di potassio. Un'altra interessante ossidazione di solfuri a solfossidi si può effettuare con sodio periodato.
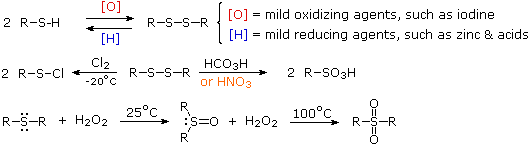 La nomenclatura dei composti dello zolfo è generalmente semplice. Il prefisso tio indica la sostituzione di un atomo di ossigeno con un atomo di zolfo. Quindi, -SH è un tiolo e C=S un tiochetone. Il prefisso tia indica la sostituzione di un atomo di carbonio in una catena o un anello con uno di zolfo, sebbene un singolo zolfo tipo-etere sia di solito chiamato solfuro. Per esempio, il C2H5SC3H7 è l'etil propril solfuro ed il C2H5SCH2SC3H7 può essere chiamato 3,5-ditiaottano. I solfonati sono esteri dell'acido solfonico e i sultoni sono gli equivalenti dei lattoni. Altri nomi sono indicati nella tabella sopra.
La nomenclatura dei composti dello zolfo è generalmente semplice. Il prefisso tio indica la sostituzione di un atomo di ossigeno con un atomo di zolfo. Quindi, -SH è un tiolo e C=S un tiochetone. Il prefisso tia indica la sostituzione di un atomo di carbonio in una catena o un anello con uno di zolfo, sebbene un singolo zolfo tipo-etere sia di solito chiamato solfuro. Per esempio, il C2H5SC3H7 è l'etil propril solfuro ed il C2H5SCH2SC3H7 può essere chiamato 3,5-ditiaottano. I solfonati sono esteri dell'acido solfonico e i sultoni sono gli equivalenti dei lattoni. Altri nomi sono indicati nella tabella sopra.
3. Ossidazione di alcoli con DMSOLa conversione di alcoli primari e secondari ad aldeidi e chetoni è un'importante reazione che, nella sua forma più semplice, può essere considerata una deidrogenazione (perdita di H2). Fornendo una fonte di ossigeno per fissare l'idrogeno prodotto come acqua, il processo di deidrogenazione endotermica può essere convertito in uno esotermicoo più favorevole. Una fonte di ossigeno che si è dimostrata efficace per l'ossidazione degli alcoli è un semplice solvente solfossido, il DMSO. La reazione è facile dal punto di vista operativo: una soluzione in DMSO dell'alcole è trattata con uno dei diversi reagenti elettrofili deidratanti (E). L'alcole è ossidato; il DMSO è ridotto a dimetil solfuro; e l'acqua è trasferita dall'elettrofilo. A causa della natura esotermica della reazione, di solito si opera a -50 °C o anche meno. Co-solventi come il cloruro di metilene o il THF sono necessari, poiché il DMSO puro congela a 18 °C. La reazione del cloruro di ossalile con DMSO può generare il clorodimetilsolfonio cloruro che quindi ossida l'alcole (ossidazione di Swern). In alternativa, un meccanismo generale plausibile per questa interessante ed utiler reazione è disegnato sotto.
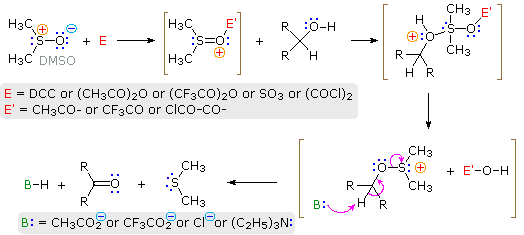
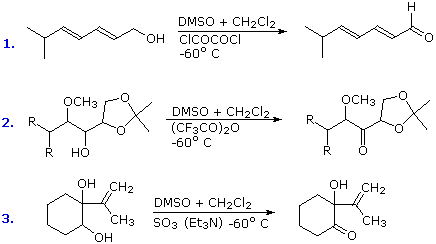
Dato che sono stati usati così tanti elettrofili diversi per fare questa ossidazione, è difficile presentare un meccanismo generale univoco. La maggior parte degli elettrofili sono buoni reagenti alchilanti, così è ragionevole aspettarsi una acilazione iniziale dell'ossigeno del solfossido. Il carattere elettrofilo dell'atomo di zolfo è aumentato dall'acilazione. Quindi segue il legame dello zolfo all'atomo di ossigeno dell'alcole. I rimanenti passaggi sono di eliminazione, in modo simile a quelli proposti per altre ossidazioni di alcoli. In alcuni casi viene aggiunta trietilamina per fornire una base addizionale. Tre esempi di queste ossidazioni con DMSO sono dati nello schema seguente. Notate che questa procedura di ossidazione è molto blanda e tollera una varietà di altri gruppi funzionali, inclusi quelli aventi atomi di azoto e zolfo ossidabili.
4. Nucleofilicità dei composti del fosforo
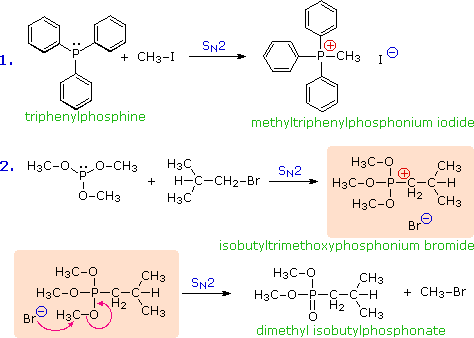
Gli analoghi al fosforo delle amine sono chiamati fosfine. La chimica delle fosfine e dei relativi esteri fosfito è dominata dalla loro forte nucleofilicità e dal loro carattere riducente. La nucleofilicità del fosforo trivalente risulta nella rapida formazione di sali di fosfonio quando tali composti vengono trattati con alogenuri alchilici reattivi. Per esempio, sebbene la delocalizzazione di risonanza del doppietto elettronico nella trifenilamina, (C6H5)3N, lo renda relativamente non reattiva in reazioni SN2, il corrispondente composto del fosforo, la trifenilfosfina, subisce una reazione rapida ed esotermica a dare un sale di fosfonio, come mostrato sotto nella prima equazione. Gli esteri fosfito reagiscono in modo simile, ma i risultanti sali di fosfinio (riquadro ombreggiato) sono spesso instabili, e per riscaldamento portano ad esteri dialchil fosfonato attraverso una seconda reazione di SN2 (equazione 2 sotto).
5. Stati di ossidazione dei composti del fosforo
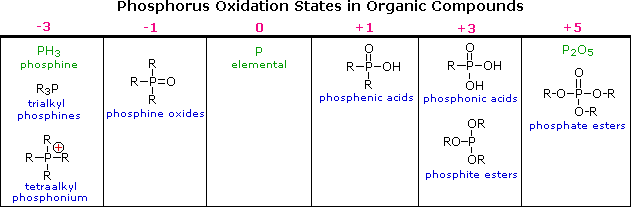
La differenza negli stati di ossidazione tra azoto e fosforo è meno pronunciata di quella tra ossigeno e zolfo. I composti organofosforo che hanno stati di ossidazione del fosforo che vanno da –3 a +5, sono mostrati nella tabella sotto, sono ben noti (alcuni semplici composti inorganici sono mostrati in verde). Come nel caso dello zolfo, i doppi legami P=O disegnati in alcune delle formule non consistono degli abituali orbitali sigma & pi ritrovati nei doppi legami carbonio-carbonio. Il fosforo è un elemento del terzo periodo, ed ha cinque orbitali 2d vuoti che possono essere usati per il legame p-d in un modo simile a quello p-p (π). In questo modo il fosforo può espandere l'ottetto di valenza tipo-argo di due elettroni (fosfin ossidi).
6. Composti del fosforo come agenti riducenti
Il fosforo trivalente è facilmente ossidato. Rispetto all'ammoniaca e alle amine, la fosfina ed i suoi mono e dialchil derivati sono piroforici, poiché bruciano con una fiamma intensa a contatto con l'ossigeno nell'aria. L'affinità del fosforo trivalente per l'ossigeno (e lo zolfo) è stata impiegata in molti sistemi reattivi, tre dei quali sono mostrati qui. Il trifenilfosfin ossido prodotto nelle reazioni 1 & 3 è un composto polare molto stabile, e nella maggior parte dei casi è facilmente rimosso dall'altro prodotto di reazione. La reazione 2 è una formulazione generale dell'utile processo di Corey-Winter per la conversione di glicoli vicinali ad alcheni.
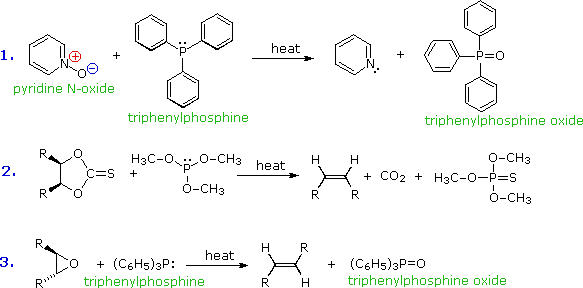
La trifenilfosfina è anche ossidata dagli alogeni, e con il bromo porta a dibromotrifenilfosforano, un composto cristallino tipo-sale, utile per la conversione di alcoli ad alchil bromuri. Come alcuni esempi precedenti, la formazione del trifenilfosfin ossido nel passaggio irreversibile SN2 fornisce una driving force termodinamica per la reazione.
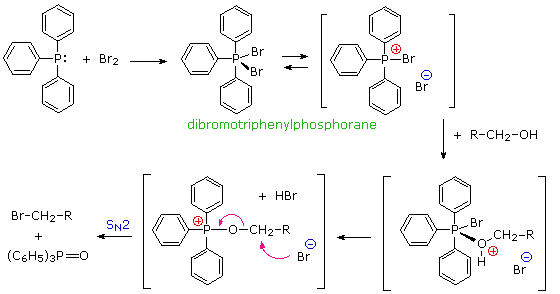
1. Preparazione di ilidi
È stato notato che gli ossidi di fosforo e zolfo dipolari sono stabilizzati tramite legame p-d. Questo può essere illustrato tramite una descrizione della risonanza, come mostrato qui.
Questa stabilizzazione legante si estende ai carboni in alfa ai centri fosfonio e solfonio, e le basi coniugate zwitterioniche derivata da tali cationi sono note come ilidi. Approssimazioni delle pKa per alcuni precursori di ilidi e relativi composti sono fornite nella seguente tabella. The acidic hydrogen atoms are colored red. Per convenzione, le pKa sono di solito aggiustate per conformarsi al solvente standard acqua; comunque, in pratica, le misure di acidi deboli sono necessariamente fatte in solventi non acquosi come il DMSO (dimetil solfossido). I numeri in verde nella tabella rappresentano le misure in DMSO, e sebbene queste siano maggiori delle approssimazioni acquose, l'ordine relativo è immutato. Notate che il DMSO stesso l'acido più debole di quelli mostrati.

Alcune preparazioni caratteristiche delle ilidi sono mostrate sotto.
Basi davvero forti, come il butil litio, sono richieste per la completa formazione delle ilidi. Il sodio idruro (NaH), altra base forte, è insolubile nella maggior parte dei solventi, ma la sua reazione con DMSO (l'acido più debole nella tabella) genera una forte base coniugata, CH3S(=O)CH2– Na+, nota come sodio dimsile. Questa base solubile è ampiamente usata per la generazioni di ilidi in soluzione di DMSO.
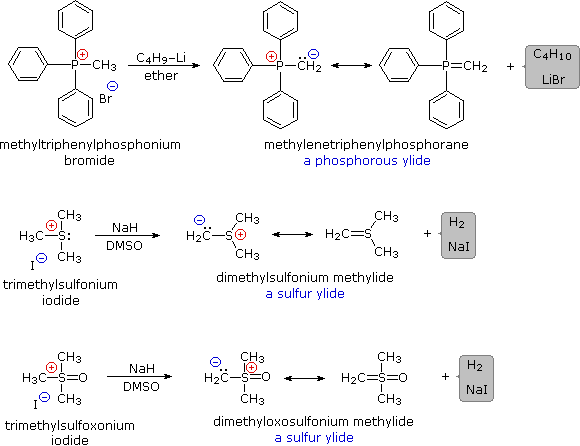
Le ilidi mostrate qui sono tutte basi forti. Come altri reagenti organici fortemente basici, esse sono protonate da acqua ed alcoli, e sono sensibili all'ossigeno. L'acqua decompone gli alchilidenfosforani ad idrocarburi e fosfin ossidi, come mostrato. L'ossigeno scinde queste ilidi in un modo simile, essendo la metà alchilidenica convertita ad un composto carbonilico.
R3P=CR'2 + H2O → [R3P(OH)–CHR'2] → R3P=O + R'2CH2
R3P=CR'2 + O2 → R3P=O + R'2C=O
2. Reazioni delle ilidi
Il più importante uso delle ilidi in sintesi proviene dalle loro reazioni con aldeidi e chetoni, che sono iniziate in ogni caso tramite un legame covalente del carbonio alfa nucleofilo verso un carbonio carbonilico elettrofilo. Le alchilidenfosforan ilidi reagiscono a dare alcheni sostituiti in una trasformazione nota come reazione di Wittig. Questa reazione è illustrata dalla prima delle tre reazioni sotto. In ogni caso il nuovo legame doppio carbonio-carbonio è colorato in blu, e l'ossigeno del reagente carbonilico è trasferito al fosforo. La reazione di Wittig tollera gli epossidi e molti altri gruppi funzionali, come dimostrato dalla reazione #1. Il centro carbanionico può anche essere sostituito, come nelle reazioni #2 & 3. Un vantaggio principale della sintesi di alcheni tramite la Wittig è che la posizione del doppio legame è assolutamente fissa, diversamente dalle miscele spesso prodotte tramite disidratazione di alcoli. Con semplici ilidi sostituite sono favoriti gli alcheni a stereochimica Z (reazione #2).
La quarta equazione mostra una reazione caratteristica di una ilide di zolfo. Ancora, l'iniziale legame carbonio-carbonio è colorato in blu, ma i passaggi successivi portano ad un epossido come prodotto piuttosto che ad un alchene.
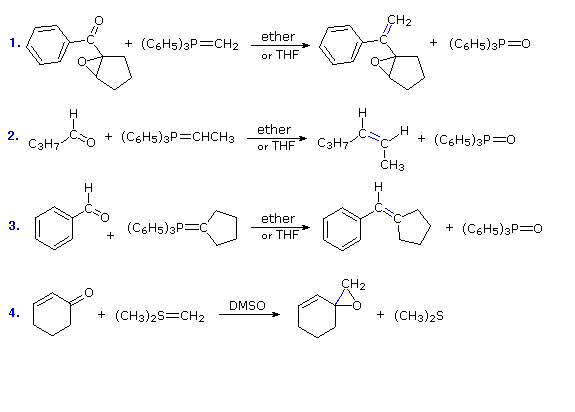
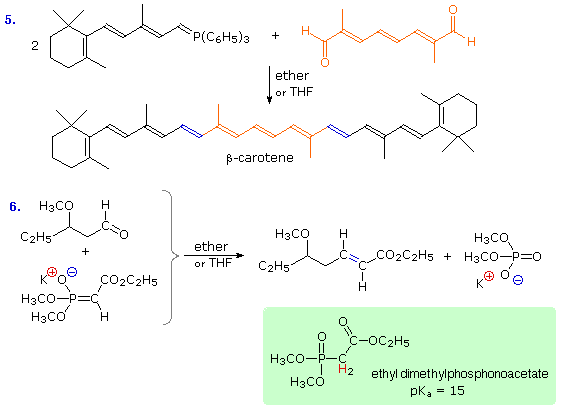
Altri due esempi di reazioni tipo-Wittig possono essere viste nel secondo schema sopra. La reazione #5 illustra una doppia reazione di Wittig, che usa un reagente dialdeidico (colorato in arancione). A causa della addizionale stabilizzazione allilica dell'ilide, i nuovi doppi legami (colorati in blu) hanno una configurazione E, in contrasto con la configurazione Z favorita da ilidi non stabilizzate (equazione 2). La reazione #6 mostra una sintesi correlata che impiega una base enolato fosfonato come nucleofilo. Questa è nota come reazione di Horner-Wadsworth-Emmons. Qui, come con la Wittig, la formazione del legame fosforo ossigeno stabile nel fosfato prodotto fornisce una driving force per la trasformazione. Ancora, la stabilizzazione del carbanione tipo-ilide porta ad una configurazione E del doppio legame prodotto. Questi cambiamenti notevoli ed utili possono essere spiegati dal meccanismo mostrato sopra.
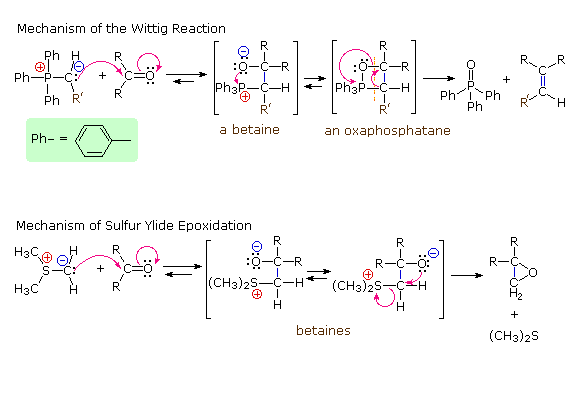
Dopo la iniziale formazione del legame carbonio-carbonio, sono stati identificati due intermedi per la reazione di Wittig, una specie dipolare a separazione di carica nota come betaina e un eterociclo a quattro membri noto come ossafosfatano. La scissione dell'ossafosfatano ad alchene e fosfin ossido è esotermica ed irreversibile. A seconda della stabilità dell'ilide di partenza, la betaina può formarsi reversibilmente e questo influenzerà alla fine la stereochimica dell'alchene prodotto.
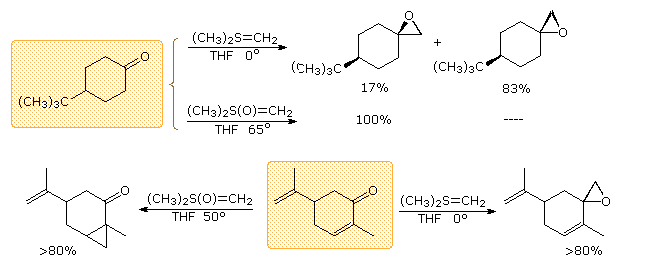
Rispetto alle ilidi di fosforo e relativi reagenti, le reazione delle ilidi di zolfo con composti carbonilici di solito non portano a specie cicliche a quattro termini analoghe agli ossafosfatani. Il cammino di reazione favorito è quindi un processo SN2 interno che porta ad un epossido come prodotto. Lo zolfo esce come dimetile solfuro.
Esempi aggiuntivi delle reazioni delle ilidi di zolfo, che illustrano le differenze nella reattività del dimetilsolfonio metiluro e dimetilossosolfonio metiluro, sono dati nello schema che segue. Dei due, l'ossosolfonio ilide è la meno reattiva e si pensa addizione reversibilmente ai gruppo carbonilici, eventualmente formando il prodotto termodinamicamente favorito.