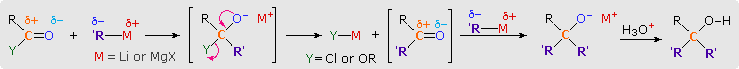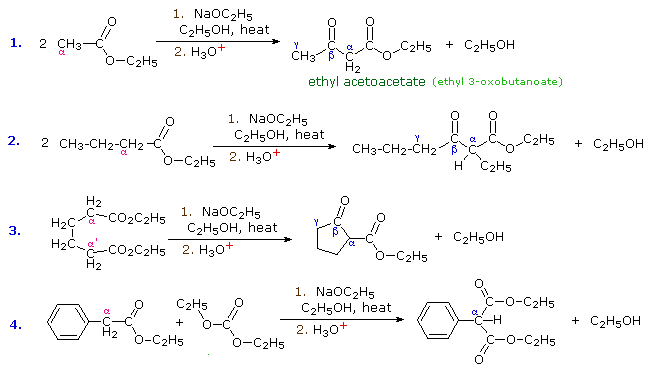quimico
2012-01-05 10:20
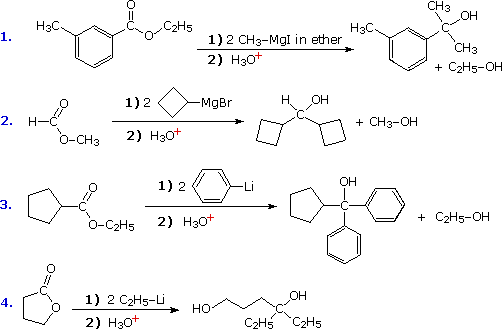
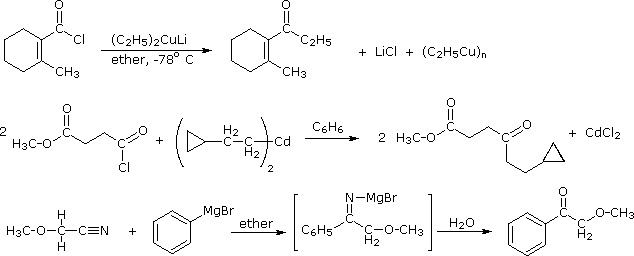
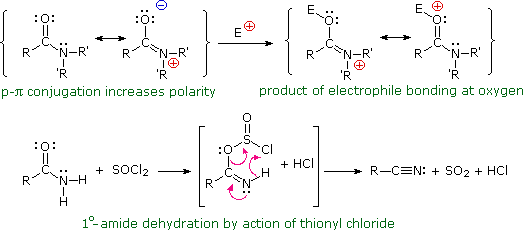
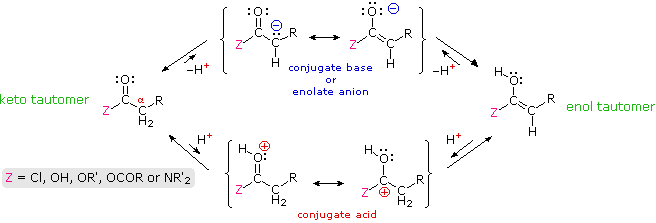
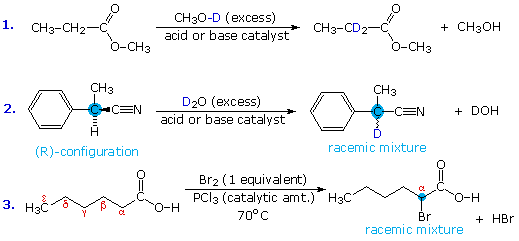
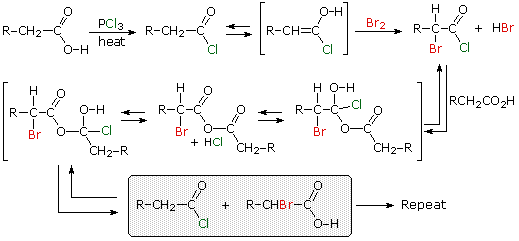
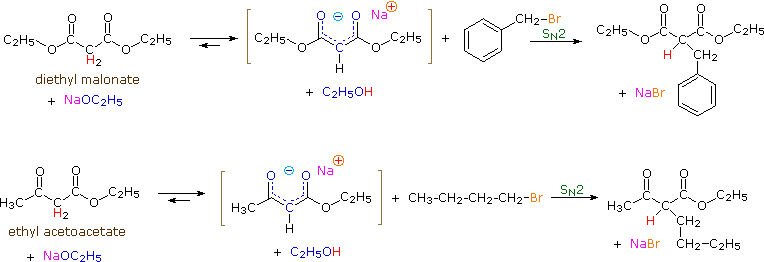
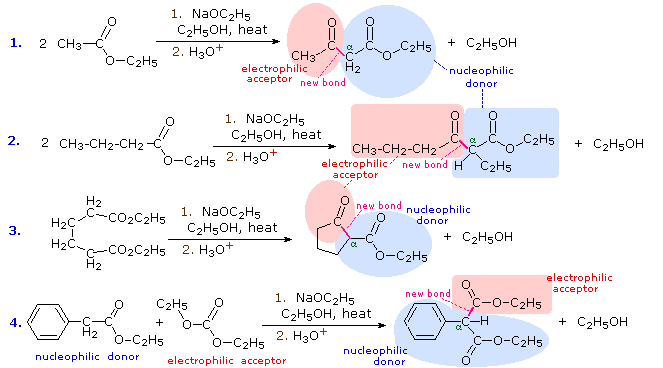
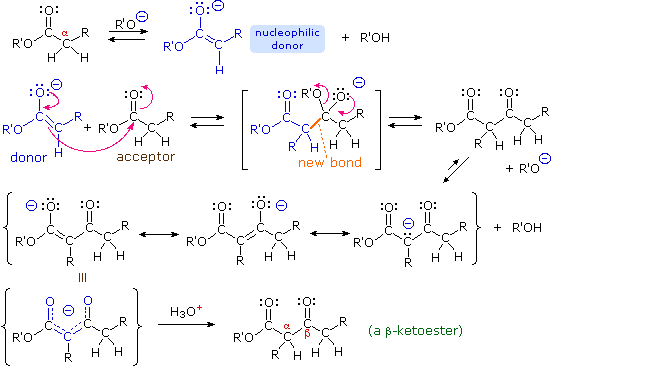
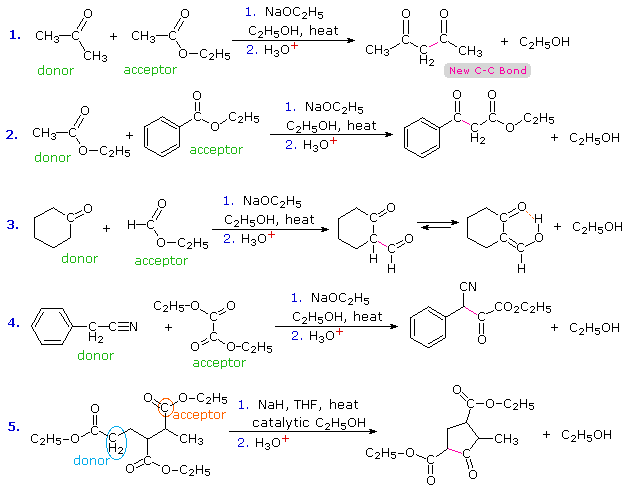
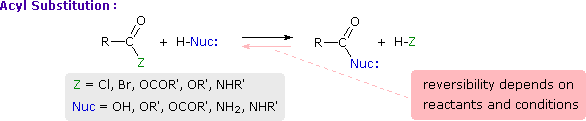
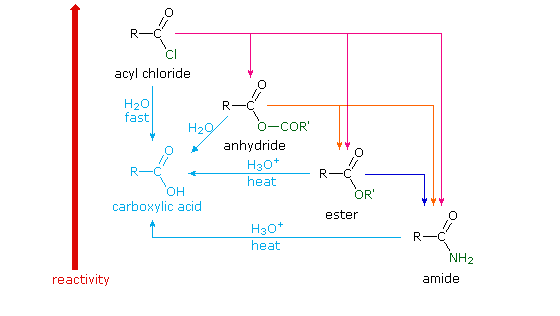
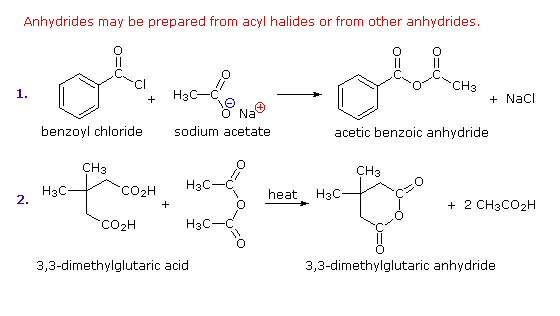
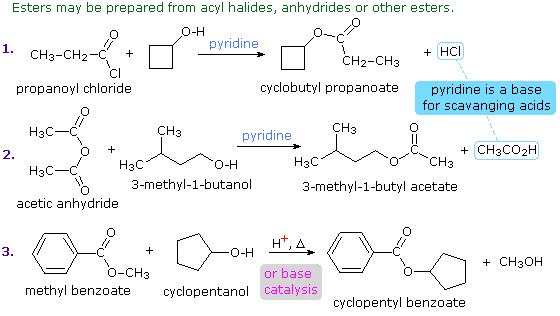
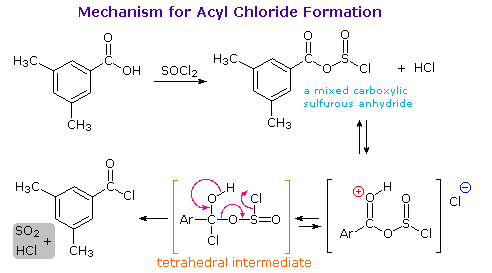
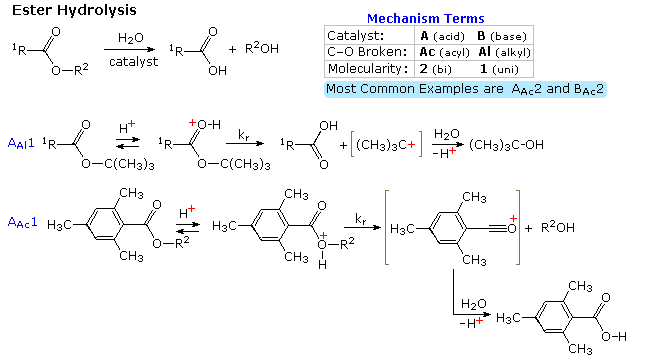
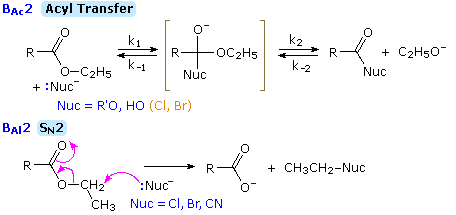
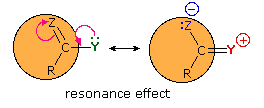
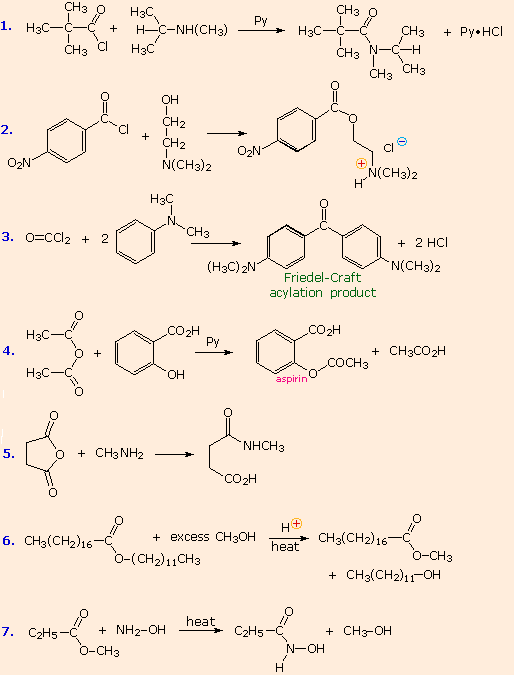
1. Background e Proprietà L'importante classe di composti organici noti come alcoli, fenoli, eteri, amine ed alogenuri è formata da gruppi alchile e/o arile legati a dei sostituenti idrossile, alcossile, amino e alogeno rispettivamente. Se questi stessi gruppi funzionali sono attaccati ad un gruppo acile (RCO–) le loro proprietà vengono sostanzialmente mutate, ed essi sono designati come derivati degli acidi carbossilici. Gli acidi carbossilici hanno un gruppo idrossile legato ad un gruppo acile, e i loro derivati funzionali sono preparati per sostituzione del gruppo idrossile con sostituenti, quali alogeno, alcossile, amino e acilossi. La seguente tabella elenca alcuni derivati rappresentativi ed i loro b.p. oltre ad altre proprietà interessanti ed importanti. Anche un'aldeide ed un chetone di equivalente peso molecolare sono elencati per paragone. I b.p. sono dati per 760 torr (pressione atmosferica), e quelli elencati come intervallo sono stimati dai valori ottenuti a pressioni inferiori. Come notato in precedenza, il b.p. relativamente alto degli acidi carbossilici è a causa dell'estesa dimerizzazione tramite legami ad idrogeno. Simili legami ad idrogeno avvengono tra le molecole di ammidi primarie e secondarie (ammidi aventi almeno un legame N–H), e i primi tre composti nella tabella servono come esempi di legami ad idrogeno. CH3(CH2)2CO2H acido butanoico PM: 88 b.p.: 164 ºC Solubilità in acqua: molto solubile CH3(CH2)2CONH2 butanammide PM: 87 b.p.: 216-220 ºC Solubilità in acqua: solubile CH3CH2CONHCH3 N-metilpropanammide PM: 87 b.p.: 205 -210 ºC Solubilità in acqua: solubile CH3CON(CH3)2 N,N-dimetiletanammide PM: 87 b.p.: 166 ºC Solubilità in acqua: molto solubile HCON(CH3CH2CH3 N-etil,N-metilmetanammide PM: 87 b.p.: 170-180 ºC Solubilità in acqua: molto solubile CH3(CH2)3CN pentanonitrile PM: 83 b.p.: 141 ºC Solubilità in acqua: leggermente solubile CH3CO2CHO anidride etanoica metanoica PM: 88 b.p.: 105-112 ºC Reagisce con l'acqua CH3CH2CO2CH3 metile propanoato PM: 88 b.p.: 80 ºC Solubilità in acqua: leggermente solubile CH3CO2C2H5 etile etanoato PM: 88 b.p.: 77 ºC Solubilità in acqua: moderatamente solubile CH3CH2COCl propanoile cloruro PM: 92.5 b.p.: 80 ºC Reagisce con acqua CH3(CH2)3CHO pentanale PM: 86 b.p.: 103 ºC Solubilità in acqua: leggermente solubile CH3(CH2)2COCH3 2-pentanone PM: 86 b.p.: 102 ºC Solubilità in acqua: leggermente solubile Le ultime nove voci nella tabella sopra non possono fungere da donatori di legami ad idrogeno, così i dimeri e gli aggregati legati tramite idrogeno non sono possibili. I b.p. relativamente alti delle equivalenti ammidi terziarie e dei nitrili sono probabilmente a causa della elevata polarità di questa funzioni. Inoltre, se non è presente il legame ad idrogeno, i b.p. di composti di dimensioni comparabili si correlano ragionevolmente bene con i loro momenti dipolari. 2. Nomenclatura Tre esempi di gruppi acile aventi nomi specifici sono stati notati in precedenza. Questi sono spesso usati nei nomi comuni dei composti. Nei seguenti esempi i nomi IUPAC sono codificati tramite colori, ed i nomi comuni sono dati tra parentesi. • Esteri: Il gruppo alchile è nominato per primo, seguito da un nomer derivato per il gruppo acile, il suffisso oico o ico nel nome dell'acido è sostituito da ato. Ad esempio il CH3(CH2)2CO2C2H5 è l'etile butanoato (o etile butirrato). Gli esteri ciclici sono chiamati lattoni. Una lettera greca identifica la posizione dell'ossigeno sulla catena alchilica rispetto al gruppo carbonilico carbossile. • Alogenuri acilici: Il gruppo acile è nominato per primo, seguito dal nome dell'alogeno come parola separata. Ad esempio il CH3CH2COCl è il propanoile cloruro (o propionile cloruro). • Anidridi: La parola separata "anidride" è usata per prima, seguita dal nome dell'acido o degli acidi correlati. Ad esempio il (CH3(CH2)2CO)2O è l'anidride butanoica & CH3COOCOCH2CH3 è l'anidride etanoica propanoica (o anidride acetica propionica). • Ammidi: Il nome dell'acido correlato è usato per primo e il suffisso dell'acido oico o ico è sostituito da ammide (solo per ammidi primarie). Ad esempio CH3CONH2 è l'etanammide (o acetammide). Le ammidi secondarie & terziarie hanno sostituenti alchilici sull'atomo di azoto. Queste sono designate tramite il termine o i termini "N-alchil" all'inizio del nome. Ad esempio CH3(CH2)2CONHC2H5 è l'N-etilbutanammide; & HCON(CH3)2 è l'N,N-dimetilmetanammide (o N,N-dimetilformammide). Le ammidi cicliche sono chiamate lattami. Un lettera greca identifica la posizione dell'azoto sulla catena alchilica rispetto al gruppo carbonilico carbossile. • Nitrili: I nitrili aciclici semplici sono nominati aggiungendo nitrile come suffisso al nome dell'alcano corrispondente (stesso numero di atomi di carbonio). La numerazione della catena inizia con il carbonio del nitrile. Comunemente, le terminazioni acido -oico o -ico dell'acido carbossilico corrispondente sono sostitute da onitrile. Un sostituente nitrile, ad esempio su un anello, è chiamato carbonitrile. Ad esempio (CH3)2CHCH2C≡N è il 3-metilbutanonitrile (o isovaleronitrile).
I seguenti utenti ringraziano quimico per questo messaggio: jobba
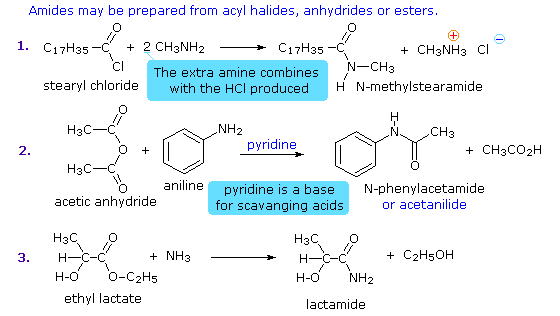
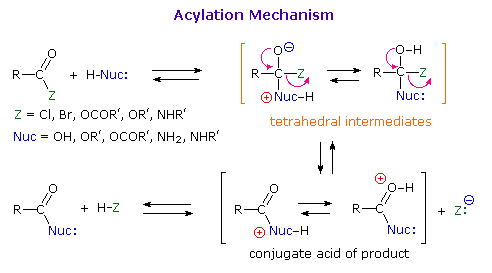
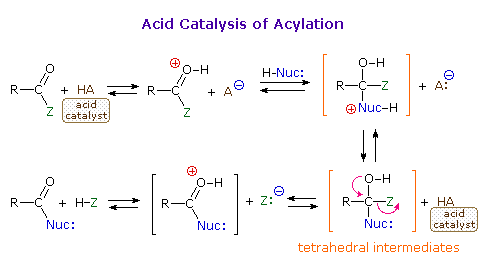
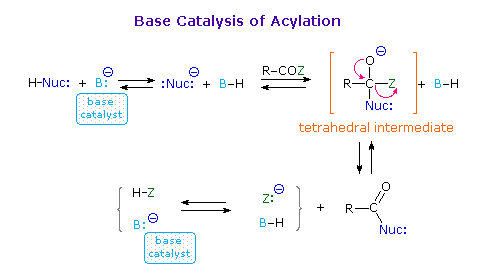
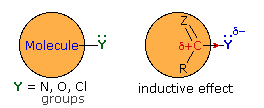
 RCONR'3+Cl– (un sale acilammonio)
RCONR'3+Cl– (un sale acilammonio) (in presenza di OH–/Δ) R–CO–NH(CH3) + H2O (in presenza di H+/Δ)
(in presenza di OH–/Δ) R–CO–NH(CH3) + H2O (in presenza di H+/Δ)  R–CO2H + CH3NH3+
R–CO2H + CH3NH3+
 R2C=C=O (un dialchilchetene) + R'3NH+ Cl–
R2C=C=O (un dialchilchetene) + R'3NH+ Cl–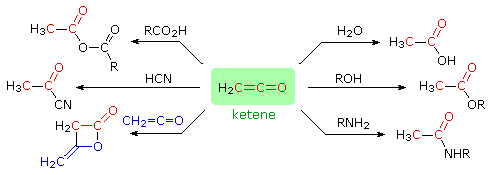
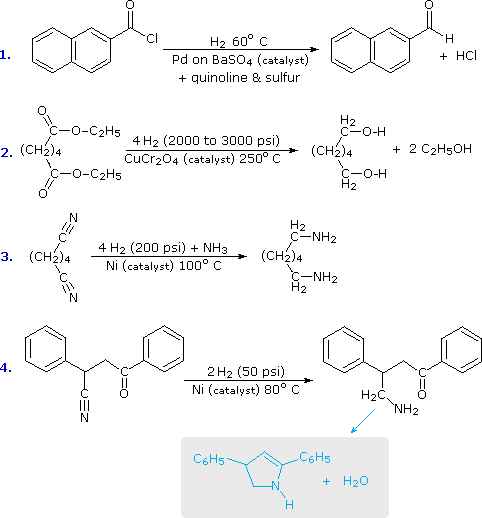
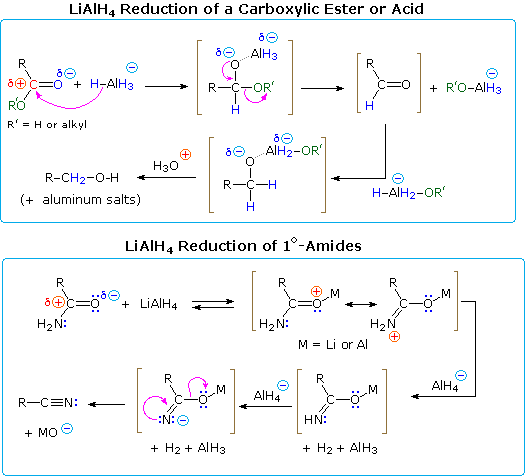 Il LAH idruro riduce i nitrili ad amine primarie, come nella seguente equazione. Una iniziale addizione di idruro all'atomo di carbonio elettrofilo del nitrile genera il sale di un'immina intermedia. Questa è seguita da un secondo trasferimento di idruro, e il risultante sale metallico dell'amina è idrolizzato ad un'amina primaria. Questo metodo fornisce una utile alternativa alla riduzione catalitica dei nitrili, descritta sopra, quando sono presenti funzioni alchene o alchino.
Il LAH idruro riduce i nitrili ad amine primarie, come nella seguente equazione. Una iniziale addizione di idruro all'atomo di carbonio elettrofilo del nitrile genera il sale di un'immina intermedia. Questa è seguita da un secondo trasferimento di idruro, e il risultante sale metallico dell'amina è idrolizzato ad un'amina primaria. Questo metodo fornisce una utile alternativa alla riduzione catalitica dei nitrili, descritta sopra, quando sono presenti funzioni alchene o alchino.
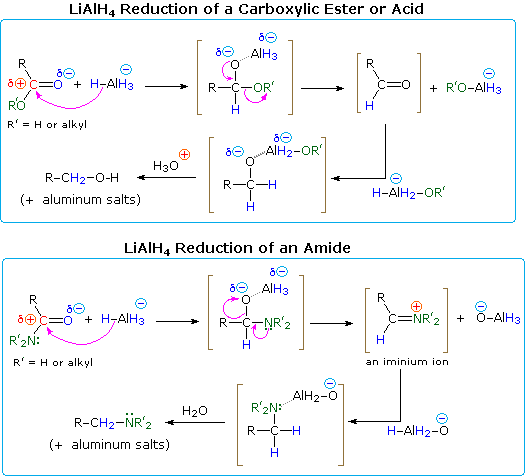
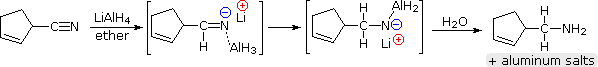 Diversamente dall'utilità del LAH nel ridurre diversi derivati degli acidi carbossilici, il sodio boroidruro è raramente schelto per questo fine. Primo, il NaBH4 è spesso usato in solventi idrossilici (acqua ed alcoli), e questi reagirebbero con i cloruri acilici e le anidridi. Inoltre, esso è moderatamente solubile in solventi relativamente non polari, particolarmente a basse temperature. Secondo, il NaBH4 è molto meno reattivo del LAH, dato che fallisce anche nel ridurre le ammidi e gli acidi (essi formano sali carbossilato), e dato che riduce gli esteri molto lentamente.
Dato che relativamente pochi metodi esistono per la riduzione degli acidi carbossilici ad aldeidi, sarebbe utile modificare la reattività e la solubilità del LAH in modo da permettere che le riduzioni parziali di questo tipo vengano realizzate. L'approccio più fruttuoso a questo scopo è stato l'attacco di gruppi alcossi o alchile sull'alluminio. Questo non solo modifica la reattività del reagente come donatore di idruro, ma aumenta anche la sua solubilità in solventi non polari. Due reagenti di questo tipo verranno menzionati qui; l'atomo di idruro reattivo è colorato in blu.
Litio tri-t-butossialluminoidruro (LtBAH), LiAl[OC(CH3)3]3H : Solubile in THF, diglyme & etere.
Diisobutilalluminio idruro (DIBAH o DIBAL-H), [(CH3)2CHCH2]2AlH : Solubile in toluene, THF & etere.
Oguno di questi reagenti porta con sé un equivalente di idruro. Il primo (LtBAH) è un metallo idruro complesso, ma il secondo è semplicemente un derivato alchilico dell'alluminio idruro. In pratica, entrambi i reagenti sono usati in quantità equimolari, e di solito a temperature al di sotto degli 0 °C. I seguenti esempi illustrano come le aldeidi possano essere preparate dai derivati degli acidi carbossilici tramite l'attenta applicazione di questi reagenti. Una temperatura di -78 °C è facilmente mantenuta tramite l'uso del ghiaccio secco (CO2 solida) come refrigerante.
Diversamente dall'utilità del LAH nel ridurre diversi derivati degli acidi carbossilici, il sodio boroidruro è raramente schelto per questo fine. Primo, il NaBH4 è spesso usato in solventi idrossilici (acqua ed alcoli), e questi reagirebbero con i cloruri acilici e le anidridi. Inoltre, esso è moderatamente solubile in solventi relativamente non polari, particolarmente a basse temperature. Secondo, il NaBH4 è molto meno reattivo del LAH, dato che fallisce anche nel ridurre le ammidi e gli acidi (essi formano sali carbossilato), e dato che riduce gli esteri molto lentamente.
Dato che relativamente pochi metodi esistono per la riduzione degli acidi carbossilici ad aldeidi, sarebbe utile modificare la reattività e la solubilità del LAH in modo da permettere che le riduzioni parziali di questo tipo vengano realizzate. L'approccio più fruttuoso a questo scopo è stato l'attacco di gruppi alcossi o alchile sull'alluminio. Questo non solo modifica la reattività del reagente come donatore di idruro, ma aumenta anche la sua solubilità in solventi non polari. Due reagenti di questo tipo verranno menzionati qui; l'atomo di idruro reattivo è colorato in blu.
Litio tri-t-butossialluminoidruro (LtBAH), LiAl[OC(CH3)3]3H : Solubile in THF, diglyme & etere.
Diisobutilalluminio idruro (DIBAH o DIBAL-H), [(CH3)2CHCH2]2AlH : Solubile in toluene, THF & etere.
Oguno di questi reagenti porta con sé un equivalente di idruro. Il primo (LtBAH) è un metallo idruro complesso, ma il secondo è semplicemente un derivato alchilico dell'alluminio idruro. In pratica, entrambi i reagenti sono usati in quantità equimolari, e di solito a temperature al di sotto degli 0 °C. I seguenti esempi illustrano come le aldeidi possano essere preparate dai derivati degli acidi carbossilici tramite l'attenta applicazione di questi reagenti. Una temperatura di -78 °C è facilmente mantenuta tramite l'uso del ghiaccio secco (CO2 solida) come refrigerante.
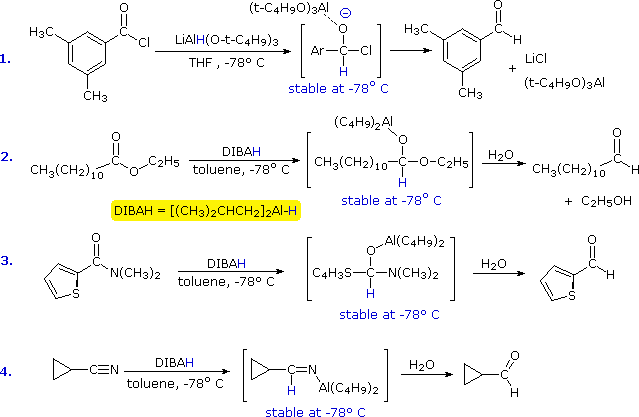
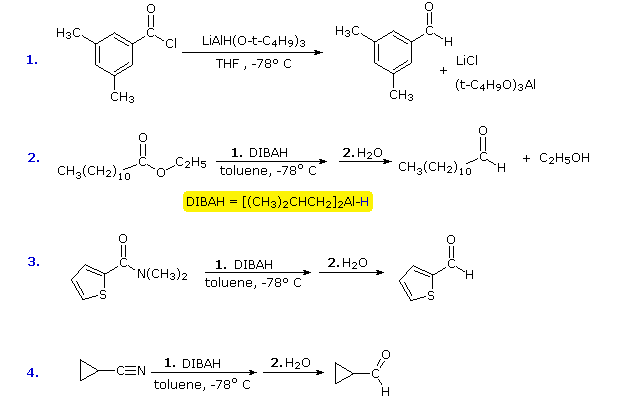 Gli intermedi ridotti che portano alle aldeidi sono mostrati negli schemi modificati sotto. Con un eccesso del reagente a temperature < 0 °C la maggior parte dei derivati degli acidi carbossilici sono ridotti ad alcoli o amine.
Gli intermedi ridotti che portano alle aldeidi sono mostrati negli schemi modificati sotto. Con un eccesso del reagente a temperature < 0 °C la maggior parte dei derivati degli acidi carbossilici sono ridotti ad alcoli o amine.
 Diborano, B2H6
Le caratteristiche riducenti del diborano (disassociato a BH3 in soluzione di etere o di THF) è stato introdotto la prima volta come reagente per le reazioni di addizione ad alcheni ed alchini. Questa rimane una applicazione primaria di questo reagente, ma effettua anche la rapida e completa riduzione degli acidi carbossilici, delle ammidi e dei nitrili. Oltre al LAH, questo reagente fornisce uno dei migliori metodi per la riduzione degli acidi carbossilici ad alcoli primari.
(1) R–CO2H + BH3 in etere
Diborano, B2H6
Le caratteristiche riducenti del diborano (disassociato a BH3 in soluzione di etere o di THF) è stato introdotto la prima volta come reagente per le reazioni di addizione ad alcheni ed alchini. Questa rimane una applicazione primaria di questo reagente, ma effettua anche la rapida e completa riduzione degli acidi carbossilici, delle ammidi e dei nitrili. Oltre al LAH, questo reagente fornisce uno dei migliori metodi per la riduzione degli acidi carbossilici ad alcoli primari.
(1) R–CO2H + BH3 in etere