L'idroformilazione di olefine (sintesi oxo) è il più vasto processo industriale basato sulla catalisi omogenea, con un scala di produzione attorno a 7x106 t/a di composti oxo e dei loro prodotti successivi.
La reazione fu brevettata nel 1938 dalla Roelen (Ruhrchemie AG), che in modo totalmente serendipity preparò la aldeide propionica da etilene e gas di sintesi in presenza di un catalizzatore etereogeneo di cobalto. Fu presto realizzato che il catalizzatore in azione era un carbonile idruro di cobalto, che derivava dalla carbonilazione riduttiva di cobalto ossido con H2 e CO.
C2H4 + CO + H2 (in presenza di HCo(CO)4, T = 90-250 °C, 100-400 bar) → CH3CH2CHO
Aldeidi con catene che vanno da C3 a C15 sono oggigiorno prodotte dalle corrispondenti olefine e successivamente convertite in amine, acidi carbossilici, e, soprattutto, alcoli primari. I più importanti prodotti oxo (ca. il 75%) sono l'1-butanolo e il 2-etilesanolo, come già accennato da chomico.
La reazione oxo formalmente implica l'addizione di H e HCO attraverso un doppio legame C=C ed è quindi definita idroformilazione.
La reattività relativa segue il seguente trend:
CH3CH2CH2CH=CH2 >> CH3CH2CH=CHCH3 > CH3CH2CH2C(CH3)=CH2 > CH3CH2CH=C(CH3)2 > (CH3)3CCH=C(CH3)2
L'idroformilazione di alcheni terminali è di importanza industriale. L'idroformilazione di dieni coniugati è impedita dalla successiva isomerizzazione e riduzione (Fell, 1977). Comunque, alcuni alcheni funzionalizzati possono essere usati:
CH2=CHCH2OH per reazione con 1. CO/H2, Rh cat. e 2. H2, Ni-Raney porta all'1,4-butandiolo (ARCO process, 1990)
L'1,4-butandiolo è un intermedio versatile e importante nella sintesi del tetraidrofurano. Il 2-etilesanolo e gli alcoli terminali superiori sono materiali grezzi nella produzione di plastificanti e detergenti.
Il ciclo sopra riportato da chomico è stato proposto da Heck e Breslow (1961) ed è stato generalmente accettato quale meccanismo per il processo di idroformilazione, sebbene i dettagli fini siano ancora oggetto di ricerca continua (cf. Markó, 1984). Meccanismi alternativi vennero proposti da Oltay (1976, studi fatti sulle condizioni attuali del processo, assunzione di un intermedio a 20 VE) e da Brown (1980, meccanismo radicalico).
La verifica sperimentale di questo meccanismo sulla base di misure cinetiche saranno estremamente difficili a causa del grande numero di variabili. Il r.d.s. è possibilmente l'addizione ossidativa di H2.
L'idroformilazione porta spesso ad una miscela di aldeidi lineari e ramificate, con una forte predisposizione verso le prime. Questa ramificazione del prodotto è conseguenza dello step di inserzione dell'alchene all'interno del legame Co-H, la cui soppressione è uno degli obiettivi dello sviluppo del catalizzatore. Quindi, catalizzatori al cobalto modificati, con l'aggiunta di un fosfano, e specialmente catalizzatori al rodio portano ad alte rese di prodotti lineari. I catalizzatori al rodio offrono un altro vantaggio ovvero condizioni di reazione miti (100 °C, 10-20 bar). Infine, nel caso di catalizzatori di rodio, la perdita di alchene per idrogenazione si attenua.
L'idroformilazione rodio-catalizzata fu sviluppata da Wilkinson (1968, 1970) e commercializzata dalla Union Carbide (1976). Questo processo, che è oggi il metodo principe per la produzione di propene, elimina alcuni degli svantaggi associati alla catalisi del cobalto ed è, in linea di principio, anche idoneo per l'applicazione di laboratorio su piccola scala.
CH3CH=CH2 + CO + H2 (H(CO)Rh(PPh3)3, T = 100 °C, 15 bar) → CH3CH2CH2CHO
La formazione preferita del prodotto lineare di idroformilazione può essere attribuito allo step di inserzione dell'alchene nel legame Rh-H, che (per ragioni steriche) avviene secondo un meccanismo anti-Markovnikov. Questa preferenza può essere ulteriormente aumentata usando ligandi al fosforo più ingombrati.
Ovviamente a causa dell'elevato costo di questi catalizzatori al rodio è essenziale un efficiente procedura di riciclo. Una soluzione elegante al problema venne trovata nel processo Rhône-Poulenc/Ruhrchemie (1984), in cui il catalizzatore omogeneo organometallico venne per la prima volta usato in fase acquosa. Venne utilizzato il catalizzatore solubile in acqua HRh(CO)(TPPTS)3, in cui il TTPTS è un trifenilfosfano che è stato solfonato tre volte in posizione meta. Le proprietà di solubilità del catalizzatore e del prodotto permettono un processo a due fasi continuo in cui il catalizzatore rimane nel mezzo di reazione acquoso e il prodotto organico (aldeide n-butirrica) è separata per semplice decantazione.
Nessuna reazione in catalisi omogenea dovrebbe essere senza una variante enantioselettiva, e questo capita anche per l'idroformilazione. Questo non deve sorprendere in vista di potenziali applicazioni di aldeidi chirali in sintesi organica. Ancora una volta, i catalizzatori con ligandi chirali vengono alla ribalta. Catalizzatori che recano come atomo centrale il platino portano a buoni valori di eccesso enantiomerico (ee), ma purtroppo l'idrogenazione e l'isomerizzazione così come un povero rappoto iso/normale rendono tale processo poco adatto. L'ultimo aspetto, comunque, è un fattore decisivo poiché solo un meccanismo Markovnikov porterà all'aldeide chirale.
Ad oggi, il migliore risultato è stato ottenuto con ligandi misti fosfano/fosfito all'atomo di rodio, la cui chiralità è basata su collegamenti α,α'-dinaftile, HRh(CO)2{(R,S)-BINAPHOS} (Nozaki, 1997).
L'idroformilazione asimmetrica sviluppata da Nozaki può essere applicata ad una vasta varietà di olefine prochirali, incluso quelle eterofunzionalizzate e i derivati 1,2-disostituiti. Questo processo è estremamente importante alla luce della vasta occorrenza di unità strutturali chirali C3 in prodotti naturali e sostanze attive.
Vi porto un esempio di idroformilazione enantioselettiva, però in presenza di un liquido ionico, un sale di imidazolio in grado di far avvenire la reazione in bifase acquosa/organica:
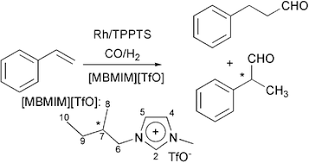
 Ciao!
Come primo, la prima cosa interessante che mi è venuta in mente è stata quella di mostrarvi un ciclo catalitico che ho disegnato per un mio prof. E' un vecchio incartapecorito, sta rifacendo le dispense di un corso di chimica industriale ed ha bisogno di una mano.
Comunque, descrivendolo brevemente, questo ciclo rappresenta l'idroformilazione del propene a butanale. Il catalizzatore, complesso idruro planare quadrato del cobalto è un realtà stato soppiantato da tre generazioni di catalizzatori più efficienti: questo è il primo cat. attivo scoperto nel 1938 circa da Otto Roelen, che ha dato vita qualche anno dopo al primo impianto della Ruhrchemie per la produzione di composti ossigenati.
Il butanale, oltre a essere ridotto all'importante solvente butanolo, può subire condensazione aldolica per dare prodotti più lunghi, come l'alcol 2-etil esanoico, usato come plastificante (se esterificato con acidi tereftalici) o come olio lubrificante (se esterificato con acidi grassi lineari, come l'acido adipico). Questi ultimi sono tutti gli oli "Synt 2000", "Synt 3456" ecc di cui fanno sempre pubblicità durante le gare di F1 haha! Il mio prof dice che questi oli sono talmente stabili che basterebbe purificarli/filtrarli dopo qualche decina di migliaia di km, per tirare così avanti per 100.000 e più km. Io ho concluso che le ditte produttrici, dal canto loro, giustamente non puntano a pubblicizzare fino in fondo queste ottime proprietà: non vogliono fare la fine della Bialetti, in fallimento perché nessuno ha più bisogno delle sue caffettiere indistruttibili!
Ciao!
Come primo, la prima cosa interessante che mi è venuta in mente è stata quella di mostrarvi un ciclo catalitico che ho disegnato per un mio prof. E' un vecchio incartapecorito, sta rifacendo le dispense di un corso di chimica industriale ed ha bisogno di una mano.
Comunque, descrivendolo brevemente, questo ciclo rappresenta l'idroformilazione del propene a butanale. Il catalizzatore, complesso idruro planare quadrato del cobalto è un realtà stato soppiantato da tre generazioni di catalizzatori più efficienti: questo è il primo cat. attivo scoperto nel 1938 circa da Otto Roelen, che ha dato vita qualche anno dopo al primo impianto della Ruhrchemie per la produzione di composti ossigenati.
Il butanale, oltre a essere ridotto all'importante solvente butanolo, può subire condensazione aldolica per dare prodotti più lunghi, come l'alcol 2-etil esanoico, usato come plastificante (se esterificato con acidi tereftalici) o come olio lubrificante (se esterificato con acidi grassi lineari, come l'acido adipico). Questi ultimi sono tutti gli oli "Synt 2000", "Synt 3456" ecc di cui fanno sempre pubblicità durante le gare di F1 haha! Il mio prof dice che questi oli sono talmente stabili che basterebbe purificarli/filtrarli dopo qualche decina di migliaia di km, per tirare così avanti per 100.000 e più km. Io ho concluso che le ditte produttrici, dal canto loro, giustamente non puntano a pubblicizzare fino in fondo queste ottime proprietà: non vogliono fare la fine della Bialetti, in fallimento perché nessuno ha più bisogno delle sue caffettiere indistruttibili! domani che sono sveglio amplio dicendo qualcosa... se mi permetti ovvio
domani che sono sveglio amplio dicendo qualcosa... se mi permetti ovvio  saluti
PS: per esser precisi ci andrebbe un 2 davanti ad HCo(CO)4
saluti
PS: per esser precisi ci andrebbe un 2 davanti ad HCo(CO)4
