quimico
2011-10-14 08:40
Ripeto qui alcune cose dette altrove. Pregherei i moderatori di pulire l'altra discussione... qui mi pare più logico parlare di organometallica. No?
I sacri testi riconoscono nella figura di Sir Edward Frankland (1825-1899) il pioniere ed inventore del termine "organometallico/a"; termine coniato da Frankland attorno al 1849, l'anno in cui ricevette il suo Ph.D. dall'Università di Marburgo.

Pietre miliari nella chimica organometallica
1760 La culla della chimica organometallica è una farmacia di Parigi. Qui lavora Cadet su un inchiostro invisibile basato su soluzioni di sali di cobalto. Per questa preparazione, usa cobalto contanimato da arsenico
As2O3 + 4 CH3COOK → [(CH3)2As]2O "liquido fumante di Cadet" - primo composto organometallico.
1827 Sale di Zeise, Na[PtCl3C2H4]: il primo complesso metallo-olefina.
1840 R.W. Bunsen continua gli studi su composti cacodilici, che rinomina "alchilarsine". La debolezza del legame As-As in molecole del tipo R2As-AsR2 lo conducono ad una serie di composti quali (CH3)2AsCN, il cui sapore (!) viene testato da Bunsen.
1849 E. Frankland, uno studente di Bunsen a Marburgo tenta la preparazione di un "radical etile" (cacodile venne anche considerato essere un radicale)
3 C2H5I + 3 Zn → (C2H5)2Zn (un liquido piroforico) + C2H5ZnI (solido) + ZnI2
Frankland è ammirevolmente specializzato nella manipolazione di composti sensibili all'aria. Usò idrogeno come gas per avere atmosfera inerte!
1852 Frankland prepara gli importanti alogenuri di alchilmercurio:
CH3I + Hg → CH3HgI (lasciò al sole la soluzione di iodometano e mercurio)
inoltre: (C2H5)4Sn, (CH3)3B (1860)
Negli anni seguenti, le reazioni con trasferimento di alchile con R2Hg e R2Zn servono nella sintesi di numerosi composti organometallici del main group.
Frankland introdusse anche il concetto di valenza.
1852 C.J. Lowig e M.E. Schweizer a Zurigo per la prima volta preparano (C2H5)4Pb da etile ioduro e un'amalgama Na/Pb. In modo simile, ottengono anche (C2H5)2Sb e (C2H5)3Bi.
1859 W. Hallawachs e A. Schafarik generano ioduri di alchilalluminio:
2 Al + 3 RI → R2AlI + RAlI2
1863 C. Friedel e J.M. Crafts preparano gli organoclorosilani:
SiCl4 + m/2 ZnR2 → RmSiCl4-m + m/2 ZnCl2
1866 J.A. Wanklyn sviluppa un metodo per la sintesi di composti di alchilmagnesio esenti da alogenuri:
(C2H5)2Hg + Mg → (C2H5)2Mg + Hg
1868 M.P. Schutzenberger ottiene [Pt(CO)Cl2], il primo complesso metallo-carbonile.
1871 D.I. Mendeleev usa composti organometallici come casi per testare la sua tavola periodica.
1890 L. Mond: Ni(CO)4, primo metallo carbonile binario, usato in un processo commerciale per raffinare Ni. Mond è il fondatore della compagnia inglese ICI (Imperial Chemical Industries).
1899 P. Barbier sostituisce Zn con Mg nella reazione di alchil ioduri.
La reazione è esplorata in maggiore dettaglio dallo studente di Barbier, V. Grignard (Premio Nobel 1912 condiviso con P. Sabatier). Sebbene meno sensibile di R2Zn, RMgX è un potente reagente alchilante.
1901 L.F.S. Kipping prepara (C6H5)2SiO, e lo chiamata difenilsilicone.
1909 W.J. Pope: formazione di (CH3)2PtI, il primo composto in cui c'è un legame σ tra metallo di transizione e parte organica.
1909 P. Ehrlich (inventore della chemioterapia e Premio Nobel 1908) introduce il Salavarsan per il trattamento della sifilide.
1917 W. Schlenk: reagenti alchillitio attraverso transalchilazione.
2 Li + R2Hg → 2 LiR + Hg
2 EtLi + Me2Hg → 2 MeLi + Et2Hg
1919 F. Hein sintetizza polifenilcromo composti, ora noti esser complessi sandwich, da CrCl3 e PhMgBr.
1922 T. Midgley e T.A. Boyd introducono Pb(C2H5)4 come additivia anti-detonanti nel gasolio.
1927 A. Job e A. Cassal preparano Cr(CO)6.
1928 W. Hieber inaugura il suo studio sistematico dei metallo carbonili:
Fe(CO)5 + H2NCH2CH2NH2 → (H2NCH2CH2NH2)Fe(CO)3 + 2 CO
Fe(CO)5 + X2 → Fe(CO)4X2 + CO
1929 F.A. Paneth genera radicali alchilici attraverso pirolisi di PbR4.
1930 K. Ziegler incoraggia un uso più intensivo degli alchillitio nella sintesi sviluppando un preparazione più semplice:
PhCH2OMe + 2 Li → PhCH2Li + MeOLi
H. Gilman: RX + 2 Li → RLi + LiX
1931 W. Heiber prepara Fe(CO)4H2, primo idruro complesso di un metallo di transizione.
1935 L. Pauling da una descrizione VB del legame nel Ni(CO)4.
1938 O. Roelen scopre il processo oxo (idroformilazione).
1939 W. Reppe inizia a lavorare su reazioni di acetileni catalizzate da metalli di transizione.
1943 E.G. Rochow: 2 CH3Cl + Si → (CH3)2SiCl2 + ... (in presenza di Cu cat., 300°C)
Questa "sintesi diretta" scatena una produzione su larga scala ed un uso dei siliconi. Il lavoro preliminare di R. Muller venne interrotto a causa della 2a G.M.
1951 M.J.S. Dewar propone una teoria del legame per i complessi di alcheni con metalli di transizione (elaborata poi da J. Chatt e L.A. Duncanson, 1953). 1951 P. Pauson (UK) e S.A. Miller (USA) ottengono il ferrocene, (C5H5)2Fe, il primo complesso a sandwich. 1952 H. Gilman prepara LiCu(CH3)2, creando quindi una nuova classe di composti sinteticamente importanti, gli organocuprati. 1953 G. Wittig sviluppa una nuova sintesi di olefine a partire da ilidi di fosfonio e composti carbonilici (Premio Nobel 1979). 1955 E.O. Fischer: sintesi razionale del bis(benzene)cromo, (C6H6)2Cr. 1955 K. Ziegler, G. Natta: poliolefine da etilene o propilene in un processo a bassa pressione che impiega catalizzatori metallici misti (alogenuri di metalli di transizione/AlR3). 1956 H.C. Brown: idroborazione (Premio Nobel 1979). 1959 J. Smith, W. Hafner: preparazione di [(C3H5)PdCl]2, creazione del campo dei complessi π-allilici di metalli di transizione. 1959 R. Criegee: stabilizzazione del ciclobutadiene tramite complessazione in [(C4Me4)NiCl2]2, verificando quindi la predizione fatta da H.C. Longuet-Higgins e L. Orgel (1955). 1960 M.F. Hawthorne prepara il dianione icosaedrico closo-borano [B12H12]2-, predetto da H.C. Longuet-Higgins (1955). 1961 D. Crowfood Hodgkins: basandosi su analisi ai raggi X di cristalli, dimostra che il coenzima della vitamina B12 contiene un legame Co-C (Premio Nobel 1964). 1963 USA: Relazioni su il dicarba-closo-borano C2B10H12 sono inviate da diversi laboratori industriali. 1963 L. Vaska: il trans-(PPh3)2Ir(CO)Cl lega reversibilmente O2. 1964 E.O. Fischer: (CO)5WC(OMe)Me, il primo complesso carbenico. 1965 G. Wilkinson, R.S. Coffey: il (PPh3)3RhCl agisce come catalizzatore omogeneo nell'idrogenazione di alcheni. 1965 R. Petit: sintesi di (C4H4)Fe(CO)3, stabilizzazione dell'antiaromatico ciclobutadiene tramite complessazione. 1965 J. Tsuji scopre il primo coupling C-C mediato da Pd. 1967 G. Wilkinson stabilizza la molecola estremamente reattiva CS (monosolfuro di carbonio) in un complesso di rodio (PPh3)2Rh(Cl)CS. 1968 A. Streitwieser: preparazione dell'uranocene, U(C8H8)2. 1969 P.L. Timms: sintesi di complessi organometallici di transizione tramite cocondensazione metallo-atomo-ligando in fase vapore (CC). 1969 A.E. Shilov scopre lo scambio H/D di alcheni con i protoni di solventi mediato da Pd(II) in soluzioni omogenee, fondando quindi il campo sempreverde dell'attivazione C-H. 1970 G. Wilkinson: alchil composti di metalli di transizione cineticamente inerti per bloccare la β-eliminazione. 1972 R.F. Heck scopre la sostituzione palladio-catalizzata di idrogeni vinilici con alogenuri arilici, benzilici e stirilici che ha in seguito dato i natali ad una delle più importanti reazioni della chimica organometallica. 1972 H. Werner: [(C5H5)3Ni2]+, il primo complesso sandwich a triplo ponte. 1973 E.O. Fischer: I(CO)4Cr(C≡R), il primo complesso carbinico. 1973 Premio Nobel a E.O. Fischer e G. Wilkinson. 1976 Premio Nobel a W.N. Lipscomb: chiarimento teorico e sperimentale della struttura e del legame nei borani. 1976 M.F. Lappert da il via al campo dei dimetalleni di elementi del main-group con la sintesi di [(Me3Si)2CH]2Sn=Sn[CH(SiMe3)2]2. 1979 H. Kopf e P. Kopf-Maier scoprono l'azione cancerostatica del titanocene dicloruro, (C5H5)2TiCl2. 1980 H. Bock: sintesi e studi del silabenzene C5H5SiH in fase gas (isolamento di matrice: G. Maier, 1982). 1981 R. West: (Mes)2Si=Si(Mes)2 (Mes = 1,3,5-trimetilbenzil radicale), il primo composto stabile con un doppio legame Si-Si. 1981 Premio Nobel a R. Hoffmann e K. Fukui: concetti semiempirici sugli OM in una discussione unificata delle struttura e della reattività di molecole inorganica, organiche e organometalliche (analogia isolobale). 1981 G .Becker sintetizza tBu-C≡P, il primo composto con un triplo legame C-P. 1982 R.G. Bergman: reazioni intramolecolari di composti organometallici di metalli di transizione con alcheni (attivazione C-H). 1985 W. Kaminsky e H. Brintzinger introducono il MAO (zirconocene dicloruro/metil allumossano chirale) come nuova classe di catalizzatori per la polimerizzazione isotattica del propilene. 1986 R. Noyori sviluppa l'addizione catalica ed enantioselettiva di reagenti organozinco R2Zn a composti carbonilici. 1989 P. Jutzi: preparazione del decametilsilicocene, Cp*2Si, ove Cp* = C5Me5-. 1989 H. Schockel sintetizza AlCl(solv), che usa nello sviluppo della chimica organometallica dell'alluminio monovalente, per esempio, Cp*4Al4 (1991). 1991 W. Uhl: sintesi dell'anionico [i-Bu12Al12]2-, un cloro-alano icosaedrico. 1993 D. Milstein riporta l'inserzione di Rh nel legame C-C (attivazione C-C). 1994 S. Harder prepara il più leggero metallocene, l'anione litocene [Li(C5H5)2]-. 1995 A.H. Zewail studia la scissione del legame M-M e M-Co in Mn2CO10 in un raggio molecolare sulla scala dei femtosecondi (10-15s) attraverso un laser pulsato (Premio Nobel 1999). 1995 G. Kubas sintetizza il primo complesso σ di un silano e studia il tautomerismo con la forma idridosilile. Questa osservazione contribuisce alla comprensione del meccanismo dell'attivazione C-H. 1996 P.P. Power prepara il primo complesso germino con un triplo legame molibdeno-germanio. 1997 C.C. Cummins: l'atomo di C come ultimo ligando in un composto organometallico: [(R2N)3MoC]-, un "complesso di carbonio". 1997 G.M. Robinson sintetizza il sale Na2[ArGa≡GaAr] e postula a un triplo legame gallio-gallio per l'anione diarilgallino. (esempio estremo di protezione sterica di un elemento strutturalmente labile!) 1999 W. Ho monitora la deidrogenazione di singole molecole di etilene su un superficie di Ni(110) tramite STM (scanning tunneling microscopy) e IETS (inelastic electon tunneling spectroscopy). 2001 Premio Nobel a K.B. Sharpless, W.S Knowles, e R. Noyori per il lavoro pionieristico nel campo della catalisi enantioselettiva. 2004 E. Carmona relaziona sul decametildizincocene Cp*Zn-ZnCp*, la prima molecola con legame Zn(I)-Zn(I) non supportato. 2005 A. Sekiguchi caratterizza totalmente R-Si≡Si-R, il primo composto con un triplo legame Si-Si. 2005 Premio Nobel a Y. Chauvin, R.R. Schrock, e R.H. Grubbs per gli studi meccanicistici e orientati all'applicazione sui catalizzatori attivi nella metatesi di olefine.
I seguenti utenti ringraziano quimico per questo messaggio: Dott.MorenoZolghetti, Beefcotto87, Max Fritz, al-ham-bic, mkuw_, Franc

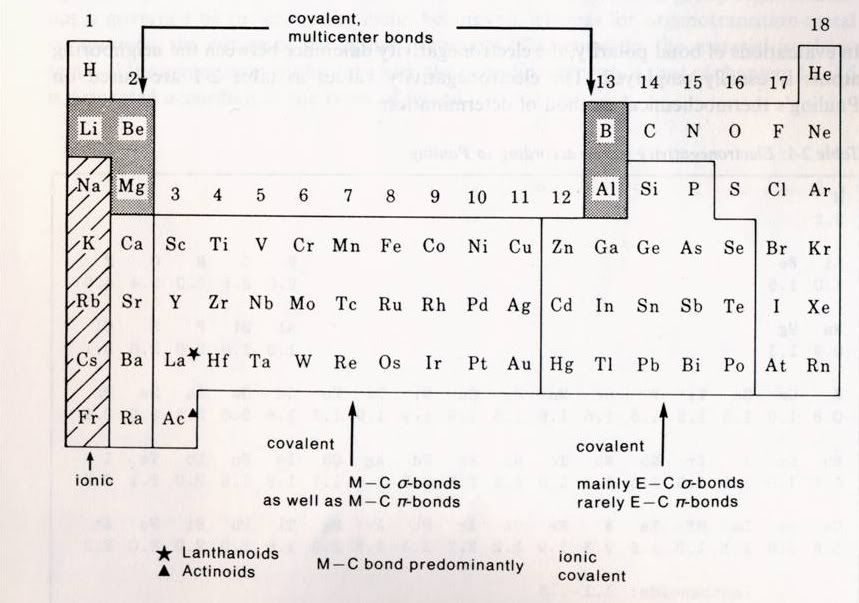
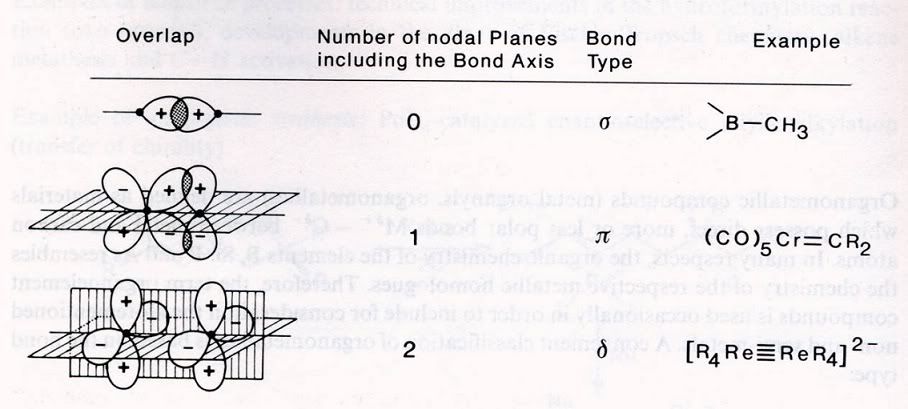

 ---
Metti pure qualcosa di preparativo, semplice magari (anche se so che non c'è praticamente niente di veramente fattibile in home-lab).
Mi son sempre piaciuti i semplici e fondamentali zinco-alchili, naturalmente mai fatti per l'infiammabilità spontanea.
---
Metti pure qualcosa di preparativo, semplice magari (anche se so che non c'è praticamente niente di veramente fattibile in home-lab).
Mi son sempre piaciuti i semplici e fondamentali zinco-alchili, naturalmente mai fatti per l'infiammabilità spontanea. sebbene il prodotto non penso sia molto salutare. Non entriamo in merito
sebbene il prodotto non penso sia molto salutare. Non entriamo in merito 
 Vedo qualcosa e magari metto ma... non prometto niente
Vedo qualcosa e magari metto ma... non prometto niente 



