quimico
2012-06-25 18:51
Visto che li ho citati nella discussione sulle reazioni multicomponente, mi pare cosa buona e giusta approfondire l'argomento.
La chimica degli isonitrili iniziò nel 1859 quando Lieke preparò l'allile isonitrile quale primo isonitrile tramite reazione dell'allile ioduro con argento cianuro (Lieke, W. "Über das Cyanallyl". Annalen der Chemie und Pharmacie (C.F. Winter'sche) 1859 112 (3): 316–321). Normalmente l'alchilazione di un cianuro di un metallo alcalino porta al nitrile, ma lo ione argento protegge l'atomo di carbonio all'estremità del cianuro.
Per il secolo seguente solo 12 isonitrili furono noti e davvero pochi tipi di reazioni erano state descritte. Quindi per un intero secolo, dal 1859 al 1958, gli isonitrili non erano facilmente disponibili, e la chimica degli isonitrili rimase una parte piuttosto vuota della chimica organica.
Gli isonitrili sono stati preparati per la prima volta indipendentemente da Gautier (A. Gautier, Ann. Chem. Pharm. 1868, 146, 119−124; Isocyanides as new isomers of cyanides are mentioned and announced previously (but not described) in: A. Gautier, Ann. Chem. Pharm. 1867, 142, 289−294) tramite reazione dell'allile ioduro con argento cianuro (alcuni testi riportano questo autore al posto di Lieke) e da Hofmann (A. W. Hofmann, Ann. Chem. Pharm. 1867, 144, 114−120) per trattamento dell'anilina con cloroformio in presenza di potassio idrossido (la nota reazione carbilaminica). A causa dell'odore estremamente sgradevole dei più semplici isonitrili (più volatili), metodi efficienti per la loro sintesi non furono sviluppati per lungo tempo, e quindi questi composti sono stati a lungo abbandonati. La chimica degli isonitrili ricevette una spinta significativa quando in letteratura apparveros metodi di sintesi sicuri per la sintesi degli isonitrili, sintesi di ampio scopo, come ad esempio la disidratazione di formammidine (I. Ugi, R. Meyr, Angew. Chem. 1958, 70, 702−703; I. Ugi, U. Fetzer, U. Eholzer, H. Knupfer, K. Offermann, Angew. Chem. 1965, 77, 492−504; Angew. Chem. Int. Ed. Engl. 1965, 4, 472−484.) e la reazione carbilaminica di amine che impiegava catalisi a trasferimento di fase (W. P. Weber, G. W. Gokel, Tetrahedron Lett. 1972, 1637−1640; W. P.
Weber, G. W. Gokel, I. K. Ugi, Angew. Chem. 1972, 84, 587; Angew. Chem. Int. Ed. Engl. 1972, 11,530−531).
L'atomo di carbonio del gruppo isonitrile spesso esibisce una reattività tipo carbene che è riflessa nella struttura di risonanza 1a. Al contrario, la struttura lineare degli isonitrili è ben rappresentata dalla struttura di risonanza dipolare 1b. Tali proprietà uniche del gruppo isonitrile, che può agire sia da elettrofilo sia da nucleofilo accoppiato alla facile disponibilità oggi di una vasta varietà di isonitrili ha reso questi composti indispensabili building blocks per la sintesi organica (M. Suginome, Y. Ito, in Science of Synthesis Vol. 19 Ed.: S.-I. Murahashi, Thieme, Stuttgart, 2004, pp. 445–530, e riferimenti citati lì).
R-N=C: (1a) ↔ R-N+≡C- (1b)
Nel 1958 Ugi ed i suoi collaboratori introdussero diversi metodi per formare isonitrili tramite disidratazione di formilamine, e quindi gli isonitrili erano facilmente disponibili.
Ugi, I. Isonitrile Chemistry. Academic Press, New York, 1971
Magari prossimamente parlerò della chimica di coordinazione di questi composti organici.
Intanto vi lascio con una chicca interessante.
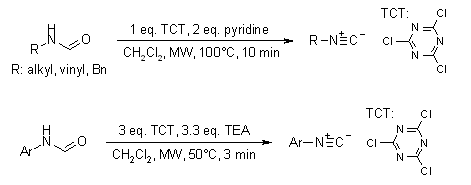
Andrea Porcheddu, Giampaolo Giacomelli e Marghareta Salaris dell'Università di Sassari (J. Org. Chem. 2005, 70, 2361) hanno descritto un metodo semplice ed efficace per la sintesi di isonitrili alifatici ed aromatici con rese elevate. Impiegando 1.3-3.0 equivalenti di un agente disidratante a basso costo quale la 2,4,6-tricloro[1,3,5]triazina (TCT) le formammidine alifatiche ed aromatiche sono state trasformate nei loro corrispondenti isonitrili con rese elevate, a 50-100 °C in 3-10 tramite microonde.
 Dimenticavo, se siete interessati vi riporto qualche metodo di sintesi pratica di questi composti.
N-Formil-2-iodoanilina
Una soluzione di o-iodoanilina (8.0 g, 36.5 mmol) ed etile formato (15 mL) in THF anidro (250 mL) è stata aggiunta goccia a goccia ad una sospensione di NaH (60% in olio minerale, 1.82 g, 45.6 mmol) in THF anidro (270 mL). La miscela risultante è stata agitata a t.a. per 24 h, e quindi la reazione è stata quenchata con acqua fredda (10 mL). I solventi sono stati rimossi a pressione ridotta, e il residuo è stata dissolto in etile acetato/acqua (400/100 mL). La fase acquosa è stata estratta con etile acetato (2 x 100 mL), gli estratti organici combinati sono stati anidrificati su Na2SO4 anidro ed evaporati. Il residuo è stato lavato a fondo con esano (3 x 100 mL) ed asciugato sotto vuoto a dare 8.84 g (98%) del composto di interesse quale solido incolore, m.p. 118 °C. Rf = 0.4 (n-Hex/EtOAc 2:1). 1H NMR (300 MHz, DMSO-d6): δ 9.50 (br s, 1 H, NH), 8.34 (s, 1 H, CHO), 7.87 (d, J = 7.7 Hz, 1 H), 7.78 (d, J = 8.1 Hz, 1 H), 7.37 (t, J = 7.3 Hz, 1 H), 6.94 (t, J = 7.3 Hz, 1 H); 13C NMR (75.5 MHz, DMSO-d6): δ 160.1 (CH), 139.0 (CH), 138.4 (C), 128.5 (CH), 126.8 (CH), 126.7 (C), 124.5 (CH); MS (70 eV, EI) m/z (%): 247.1 (48) [M+], 120.1 (100), 92.1 (50), 65.1 (60); IR (KBr): 3223, 2899, 1658 (C=O), 1583, 1572, 1524, 1463, 1435, 1394, 1281, 1240, 1163, 1151, 1017, 885, 746, 693, 644, 520, 461, 429 cm–1; Anal. Calcd per C7H6INO: C 34.03, H 2.45, N 5.67; Trovato: C. 34.23; H. 2.22; N. 5.51.
2-Iodofenil isonitrile
Ad una soluzione di N-formil-2-iodoanilina (5.11 g, 20.7 mmol) in CH2Cl2 anidro (130 mL) è stata aggiunta a 0 °C diisopropilamina (17 mL, 120 mmol), quindi goccia a goccia in 10 min POCl3 (4.4 mL, 41.4 mmol). La miscela è stata agitata a 0 °c per 15 minuti, quindi una soluzione satura di Na2CO3 (40 mL) è stata aggiunta lentamente. La miscela è stata trasferita in un imbuto separatore, diluita con diclorometano (200 mL), la fase organica lavata con una soluzione mezza saturata di Na2CO3 (100 mL) e salamoia (100 mL), quindi anidrificata su Na2SO4 anidro ed evaporata.
Il prodotto grezzo è stato purificato tramite ricristallizzazione da esano a dare 4.43 g (93%) del composto di interesse quale solido incolore, m.p. 42 °C. Rf = 0.29 (n-Hex/EtOAc 30 : 1).
1H NMR (300 MHz, CDCl3): δ 7.90 (d, J = 7.8 Hz, 1 H), 7.44–7.36 (m, 2 H), 7.14–7.09 (m, 1 H); 13C NMR (75.5 MHz, CDCl3): δ 167.4 (C), 139.6 (CH), 130.4 (2 CH), 129.0 (CH), 127.6 (C), 109.7 (C); MS (70eV, EI) m/z (%): 229.0 (100) [M+], 57.1(92), 71.1(80), 97.2(70); IR (KBr): 2123 (NC), 1459, 1434, 1042,1019, 751, 643, 432 cm–1; Anal. Calcd per C7H4IN: C 36.71, H 1.76, N 6.12; Trovato: C 36.88, H 1.88, N 5.87.
Dimenticavo, se siete interessati vi riporto qualche metodo di sintesi pratica di questi composti.
N-Formil-2-iodoanilina
Una soluzione di o-iodoanilina (8.0 g, 36.5 mmol) ed etile formato (15 mL) in THF anidro (250 mL) è stata aggiunta goccia a goccia ad una sospensione di NaH (60% in olio minerale, 1.82 g, 45.6 mmol) in THF anidro (270 mL). La miscela risultante è stata agitata a t.a. per 24 h, e quindi la reazione è stata quenchata con acqua fredda (10 mL). I solventi sono stati rimossi a pressione ridotta, e il residuo è stata dissolto in etile acetato/acqua (400/100 mL). La fase acquosa è stata estratta con etile acetato (2 x 100 mL), gli estratti organici combinati sono stati anidrificati su Na2SO4 anidro ed evaporati. Il residuo è stato lavato a fondo con esano (3 x 100 mL) ed asciugato sotto vuoto a dare 8.84 g (98%) del composto di interesse quale solido incolore, m.p. 118 °C. Rf = 0.4 (n-Hex/EtOAc 2:1). 1H NMR (300 MHz, DMSO-d6): δ 9.50 (br s, 1 H, NH), 8.34 (s, 1 H, CHO), 7.87 (d, J = 7.7 Hz, 1 H), 7.78 (d, J = 8.1 Hz, 1 H), 7.37 (t, J = 7.3 Hz, 1 H), 6.94 (t, J = 7.3 Hz, 1 H); 13C NMR (75.5 MHz, DMSO-d6): δ 160.1 (CH), 139.0 (CH), 138.4 (C), 128.5 (CH), 126.8 (CH), 126.7 (C), 124.5 (CH); MS (70 eV, EI) m/z (%): 247.1 (48) [M+], 120.1 (100), 92.1 (50), 65.1 (60); IR (KBr): 3223, 2899, 1658 (C=O), 1583, 1572, 1524, 1463, 1435, 1394, 1281, 1240, 1163, 1151, 1017, 885, 746, 693, 644, 520, 461, 429 cm–1; Anal. Calcd per C7H6INO: C 34.03, H 2.45, N 5.67; Trovato: C. 34.23; H. 2.22; N. 5.51.
2-Iodofenil isonitrile
Ad una soluzione di N-formil-2-iodoanilina (5.11 g, 20.7 mmol) in CH2Cl2 anidro (130 mL) è stata aggiunta a 0 °C diisopropilamina (17 mL, 120 mmol), quindi goccia a goccia in 10 min POCl3 (4.4 mL, 41.4 mmol). La miscela è stata agitata a 0 °c per 15 minuti, quindi una soluzione satura di Na2CO3 (40 mL) è stata aggiunta lentamente. La miscela è stata trasferita in un imbuto separatore, diluita con diclorometano (200 mL), la fase organica lavata con una soluzione mezza saturata di Na2CO3 (100 mL) e salamoia (100 mL), quindi anidrificata su Na2SO4 anidro ed evaporata.
Il prodotto grezzo è stato purificato tramite ricristallizzazione da esano a dare 4.43 g (93%) del composto di interesse quale solido incolore, m.p. 42 °C. Rf = 0.29 (n-Hex/EtOAc 30 : 1).
1H NMR (300 MHz, CDCl3): δ 7.90 (d, J = 7.8 Hz, 1 H), 7.44–7.36 (m, 2 H), 7.14–7.09 (m, 1 H); 13C NMR (75.5 MHz, CDCl3): δ 167.4 (C), 139.6 (CH), 130.4 (2 CH), 129.0 (CH), 127.6 (C), 109.7 (C); MS (70eV, EI) m/z (%): 229.0 (100) [M+], 57.1(92), 71.1(80), 97.2(70); IR (KBr): 2123 (NC), 1459, 1434, 1042,1019, 751, 643, 432 cm–1; Anal. Calcd per C7H4IN: C 36.71, H 1.76, N 6.12; Trovato: C 36.88, H 1.88, N 5.87.
I seguenti utenti ringraziano quimico per questo messaggio: Max Fritz


