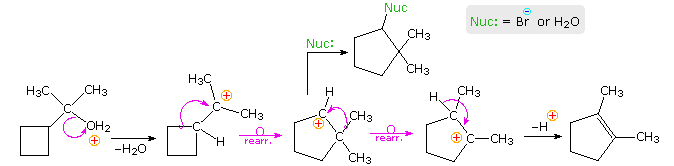
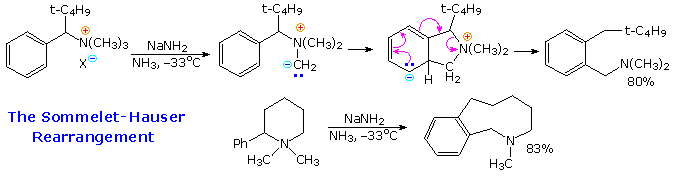
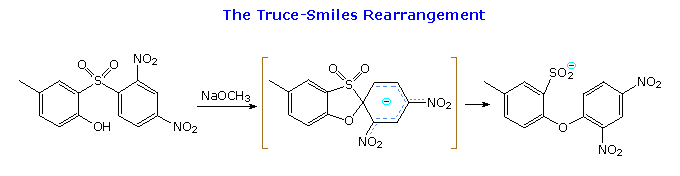
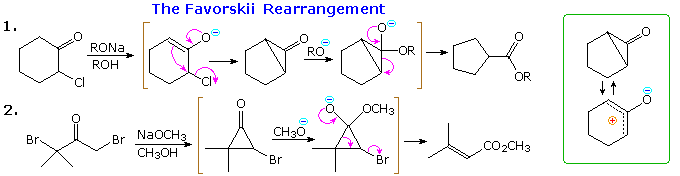
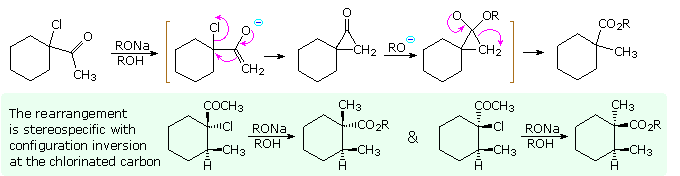
2. Riarrangiamenti di azoto cationico o elettron-deficiente
Dato che molti semplici composti azotati sono basi, essi formano cationi "onio" quando protonati. Due cationi di questo tipo sono mostrati sulla sinistra (nel riquadro blu) sotto. Poiché i cationi ammonio ed imminio hanno un azoto con un guscio elettronico di valenza pieno, un tale atomo di azoto non può fungere da sito per il legame di un nucleofilo o come estremità per la migrazione. Per un azoto cationico affinché avvenga riarrangiamento è necessario che esso abbia un guscio di valenza incompleto, e due di tali azacationi sono mostrati nel centro dello schema (riquadro rosa). Un atomo di azoto elettron-deficiente non deve essere un catione, come dimostra l'esempio del nitrene sulla destra (riquadro verde).
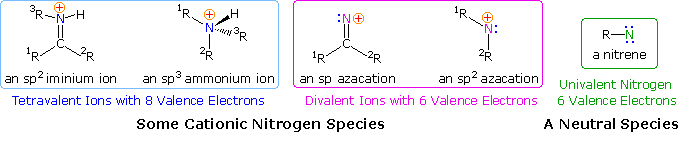
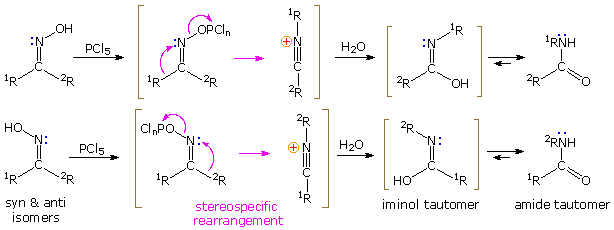
Riarrangiamento di Beckmann
Se il gruppo idrossile di una chetossima è rimosso per azione di un acido forte o di fosforo pentacloruro, seguita da idrolisi, si forma una ammide. La completa rimozione del gruppo idrossi derivatizzato e la sua coppia di elettroni leganti genererà un azacatione divalente ibridizzato sp del tipo illustrato nel precedente diagramma. Dove questo avviene, entrambi i sostituenti carboniosi (1R & 2R) dovrebbero essere i candidati per il successivo shift 1,2. In pratica, comunque, è sempre il gruppo in anti rispetto all'OH che esce che migra sull'azoto. Questa stereospecificità indica che lo shift 1,2 è concertato con la rimozione N-O, come mostrato sotto. Il risultante intermedio nitrilio N-alchilato reagirà con nucleofili (acqua) all'atomo di carbonio elettrofilo adiacente all'azoto "onio". Notare che la struttura disegnata in questo intermedio è la più favorita dei due contributi di risonanza, nella misura in cui tutti gli atomi pesanti hanno gusci di valenza pieni. Il legame di un nucleofilo all'atomo di azoto dovrebbe richiedere l'espansione del suo guscio di valenza per includere dieci elettroni, o la formazione di una specie dipolare instabile. Il prodotto iniziale dall'idratazione al carbonio è un imminolo, che immediatamente tautomerizza alla più stabile ammide. Le reazioni con PCl5 probabilmente portano ad un imminocloruro, e questo a sua volta è idrolizzato alla stessa ammide.
Il primo esempio nel seguente gruppo di reazioni è un tipico riarrangiamento di Beckmann. L'ossima dal cicloesanone ha gli identici sostituenti carbonilici e quindi esiste come singolo isomero. Il prodotto del riarrangiamento è un lattame (un'ammide ciclica), che può essere idrolizzata ad un omerga-amino acido. Questo lattame serve come importante precursore industriale per il nylon 6.
Il secondo esempio coinvolge un derivato dell'ossima con diversi sostituenti carbonilici, che esiste come coppia di stereoisomeri (syn & anti). L'isomero anti riarrangia tramite uno shift 1,2 di fenile, mentre l'isomero syn subisce uno shift 1,2 di isopropile. La prima reazione è molto più veloce dell'ultima, presumibilmente perché procede tramite un intermedio ione fenonio relativamente stabile (struttura è nel riquadro ombreggiato). Notare che il gruppo uscente picrato (2,4,6-trinitrofenolato) è un catione stabile.
L'esempio #3 è un altro caso che dimostra la stereospecificità del riarrangiamento di Beckmann. Lo shift 1,2 del sostituente o-fenolo è più veloce di quelle del gruppo fenile senza sostituenti, e l'idrossile è idealmente sito per legarsi al carbonio elettrofilo dell'intermedio. Di conseguena, il prodotto dall'isomero anti è un benzossazolo.
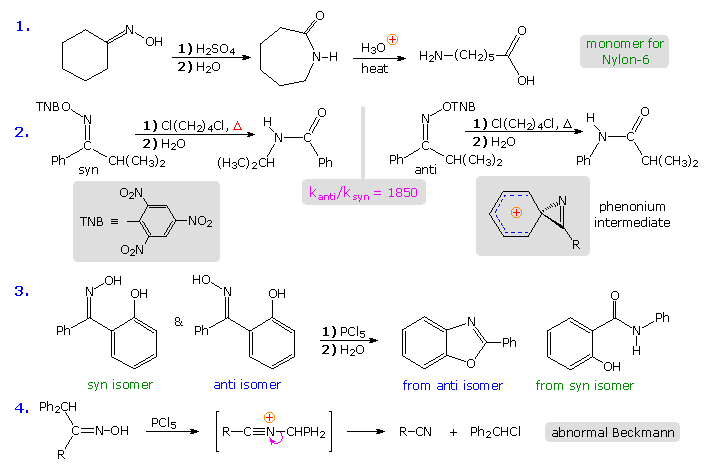
Il quarto esempio sopra mostra un'insolita divergenza nel comportamento che qualche volta accade quando i frammenti del sostituente migrante dall'intermedio, portano ad un nitrile come prodotto stabile. Questa è stata chiamata una reazione di Beckmann anomala.
la rigida configurazione del catione fenonio mostrata sopra impone una tensione strutturale che è ben dimostrata tramite le velocità di riarrangiamento di alcuni composti biciclici ad anelli fusi.
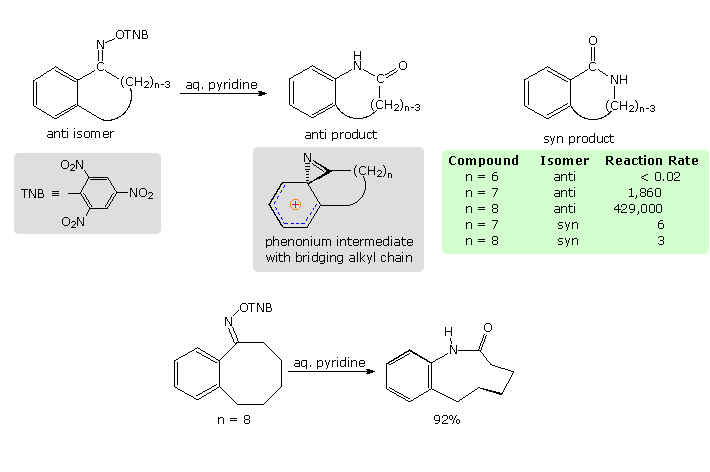
I derivati dell'ossima di fenil chetoni incorporati in anelli fusi a sei, sette ed otto membri sono stati studiati.A causa della catena di carbonio che unisce la funzione ossima al carbonio orto dell'anello benzenico, lo ione fenonio che normalmente facilita la migrazione del fenile può essere incapace di assumere la sua struttura preferita (anello a tre termini ortogonale all'anello fenile). La catena a tre carboni a ponte per n = 6 è un tale caso, ed il riarrangiamento dell'isomero anti è davvero lento. Come la lunghezza della catena che ponta aumenta, la sua tensione è meno grave, e la velocit del riarrangiamento aumenta. L'ossima del picrato ad otto membri idrolizza rapidamente, producendo un lattame a nove membri con una resa elevata.
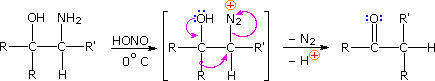
R2C=N-NH2 + HNO2 → RCONHR + N2
Riarrangiamenti tipo Beckmann possono anche essere condotti trattando idrazoni con acido nitroso, come mostrato sopra. Come regola, questa è una procedura meno apprezzata perché gli idrazoni puri sono più difficili da ottenere, e la resa del prodotto puro è inferiore.
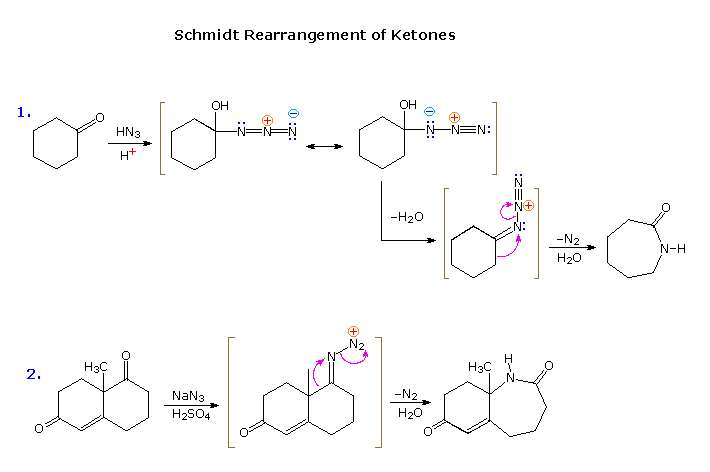
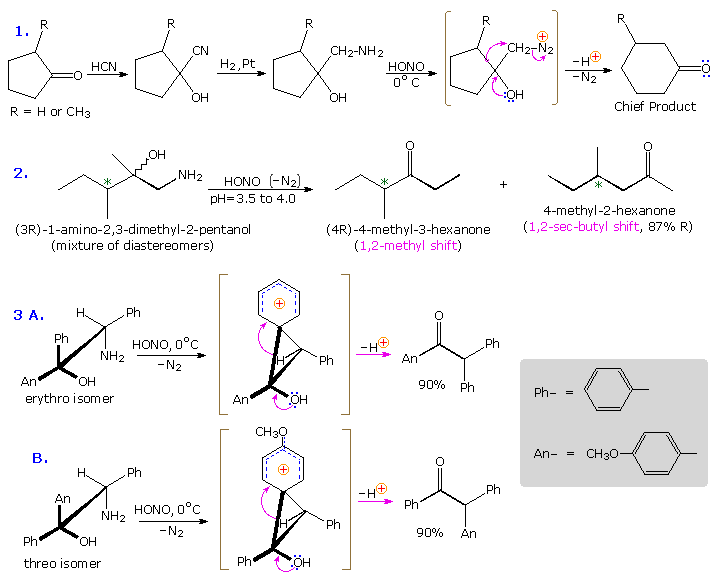
Un riarrangiamento diretto dei chetoni, evitando così la necessità di preparare un derivato, è possibile tramite una procedure nota come riarrangiamento di Schmidt. L'addizione acido catalizzata di acido idrazoico al gruppo carbonile di un chetone crea un instabile azidocarbinolo che, per disidratazione, produce lo stesso catione triazonio presumibilmente formato come intermedio nella deaminazione con acido nitrico di un idrazone. Il riarrangiamento di questa specie per rapida perdita di azoto inizia quindi un riarrangiamento di tipo Beckmann.
Sotto sono riportati due esempi del riarrangiamento di Schmidt.
Il riarrangiamento di Stieglitz
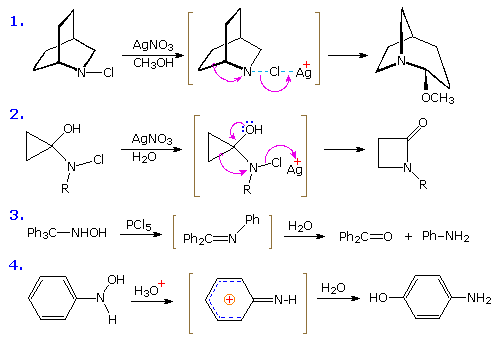
Esempi di riarrangiamento di azacationi ibridizzati sp2 sono relativamente rari comparati a quelli degli analoghi al carbonio. I materiali di partenza per generara tali azacationi sono di solito cloramine o idrossil amine. Quattro esempi di queste trasformazioni, spesso chiamate "riarrangiamenti di Stieglitz", sono mostrati sotto. Il primo esempio è simile ad un riarrangiamento di Wagner-Meerwein, ed il secondo ad un riarrangiamento pinacolic. Shifts competitivi di gruppi fenile p-sostituiti in reazioni simili all'esempio #3, dimostrano che un sostituente metossi facilita il riarrangiamento, mentre un sostituente nitro lo ritarda. Nell'ultimo esempio, un azacatione coniugato attiva l'anello benzenico alla sostituzione nucleofila, in contrasto con il solito ruolo giocato dai sostituenti di un'amina.
Riarrangiamento di acil nitreni ad isocianati
Diverse procedure utili e generali per la degradazioni di derivati di acidi carbossilici ad amine tutte comportano il riarrangiamento di un acil nitrene ad un isocianato. Sebbene l'atomo di azoto di un nitrene non ha carica formale, esso è elettron-deficiente e serve come luogo per riarrangiamenti 1,2. Come illustrato nel seguente schema, gli acil nitreni possono essere generati da diversi composti tipo ammide. Una volta formati, gli acil nitreni rapidamente riarrangiano agli isomeri isocianato relativamente stabili, i quali possono essere isolati o reagire con solventi idrossilici. La più comune applicazione di questi riarrangiamento è per la sintesi di amine. Perciò, l'addizione di acqua ad isocianati tipo chetene producono un instabile acido carbammico che decompone ad amina e diossido di carbonio. Le procedure generali per l'ottenimento di precursori nitrenici sono elencati sotto lo schema.
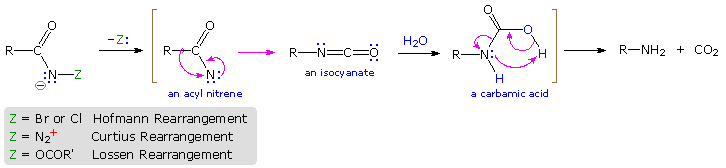
Hofmann: Ammidi primarie sono convertite a derivati N-alogenati tramite l'azione di HOX o X2 in soluzione alcalina. Un eccesso di base genera una base coniugata del prodotto.
Lossen: Un derivato dell'acido idrossamico (RCONHOH) è prodotto tramite reazione di un estere con idrossil amina. L'acido idrossamico è O-acilato e quindi convertito nella sua base coniugata.
Curtius: Un'acil azide (RCON3) è preparata in uno di due modi. (i) Reazione di un cloruro acilico con sodio azide, o (ii) Reazione di un estere con un eccesso di idrazina, seguita dalla reazione dell'acilidrazide prodotta (RCONHNH2) con acido nitroso freddo. Le acil azidi decompongono ad isocianati per riscaldamento.
Schmidt: Una variante della procedura di Curtius in cui un acido carbossilico è scaldato con acido idrazoico (HN3) e un catalizzatore acido.
Alcuni esempi di questi differenti riarrangiamenti sono mostrati nello schema sottostante. In ogni caso una lettera verde maiuscola (C, H, L o S) designa il tipo di reazione. Le prime tre reazioni illustrano il riarrangiamento di Hofmann, che è un metodo particolarmente utile per la preparazione di amine. L'ultimo esempio mostra una reazione di Curtius applicata ad un diestere tramite un intermedia bis-acilidrazide. Un approccio di Curtius alternativo per l'amina dell'esempio #2 è anche mostrato. La procedura di Curtius è particolarmente utile se l'isocianato è il prodotto desiderato, dato che la decomposizione dell'acil azide intermedia può essere condotto in assenza di solventi idrossilici che reagirebbero con l'isocianato.
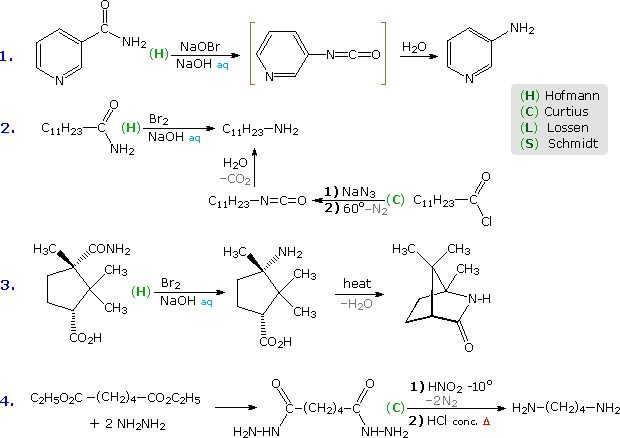
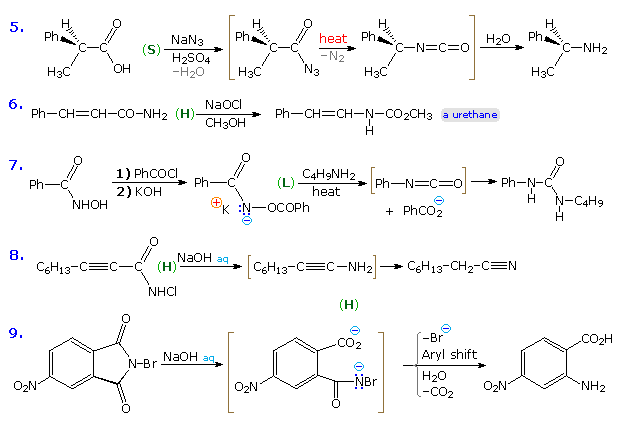
L'esempio #5 mostra una reazione di Schmidt in cui un acido carbossilico otticamente attivo è il substrato. La configurazione S del gruppo fenetile migrante è ritenuta nell'amina prodotta, confermando il carattere intramolecolare di questi riarrangiamenti. La ritenzioni di configurazione è anche osservata nei riarrangiamenti di Curtius, Lossen e Hofmann di questo tipo. Le reazioni #6 & #7 sono interessanti casi in cui l'acqua è assente durante la formazione e la rerazione dell'isocianato. Il solvente alcolico in #6 si addiziona all'isocianato per produrre un estere carbammico, noto come uretano. Diversamente dagli instabili acidi carbammici, gli uretani non decompongono e possono essere composti puri. Se l'acqua era stata il solvente, la risultante enammina primaria dovrebbe aver riarrangiato ad immina ed essersi idrolizzata ad aldeide. Nel riarrangiamento di Lossen (#7) la butil amina si addiziona all'isocianato a dare un'urea sostituita.
Il riarrangiamento di Hofmann nella reazione #8 fornisce un nuovo esempio del tautomerismo di un amina primaria acetilenica ad un nitrile. Infine, l'ultimo esempio illustra un riarrangiamento di Hofmann selettivo di una bromo-immide. La reattività del gruppo carbonile in para rispetto al gruppo elettron-attrattore nitro è aumentata rispetto all'altra immide carbonilica. Di conseguenza, l'idrolisi base catalizzata avviene qui preferenzialmente, lasciando l'acil nitrene meta alla funzione nitro.
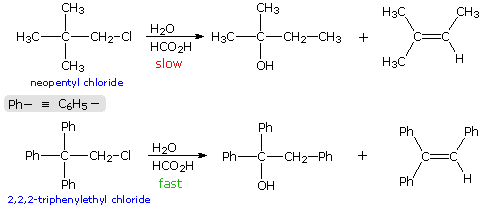
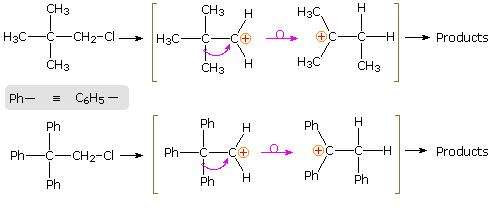
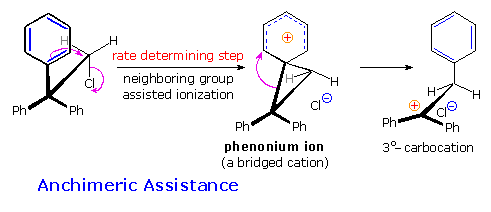
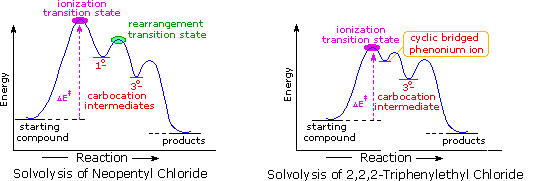
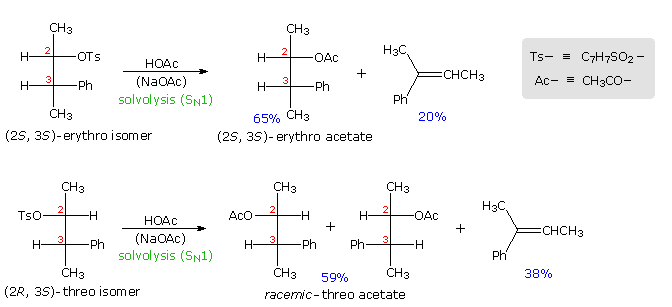
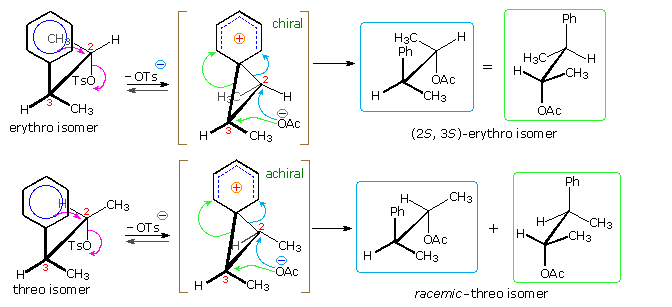
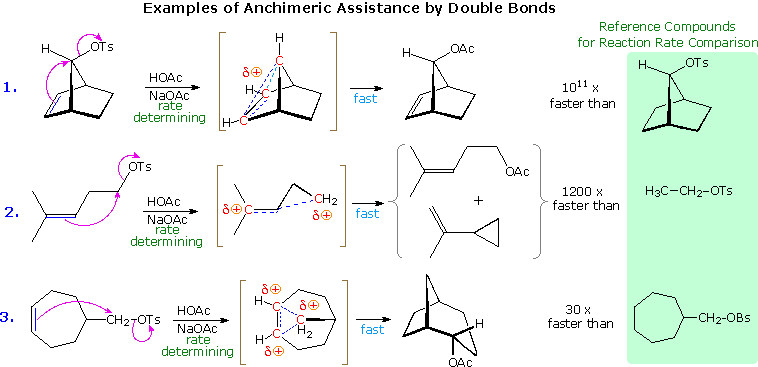 La reazione (1) è un esempio emozionante di come un doppio legame orientato in modo opportuno possa assistere la ionizzazione di un buon gruppo uscente. Non avviene alcun riarrangiamento perchè ne risulterebbero piccoli anelli tensionati.
La reazione (2) coinvoolge una molecola reagente meno rigida. La velocità di accelerazione è meno pronunciata, ma un po' di riarrangiamento è osservato. Notare che si forma un anello a tre termini adiacente al carbocatione terziario invece di un anello a quattro termini che incorpora un carbocatione secondario.
La reazione (3) mostra un caso in cui il doppio legame è ancora più lontano dal gruppo uscente. L'aumento di velocità è piccolo, ma si forma solo l'endo-acetato riarrangiato.
La reazione (1) è un esempio emozionante di come un doppio legame orientato in modo opportuno possa assistere la ionizzazione di un buon gruppo uscente. Non avviene alcun riarrangiamento perchè ne risulterebbero piccoli anelli tensionati.
La reazione (2) coinvoolge una molecola reagente meno rigida. La velocità di accelerazione è meno pronunciata, ma un po' di riarrangiamento è osservato. Notare che si forma un anello a tre termini adiacente al carbocatione terziario invece di un anello a quattro termini che incorpora un carbocatione secondario.
La reazione (3) mostra un caso in cui il doppio legame è ancora più lontano dal gruppo uscente. L'aumento di velocità è piccolo, ma si forma solo l'endo-acetato riarrangiato.
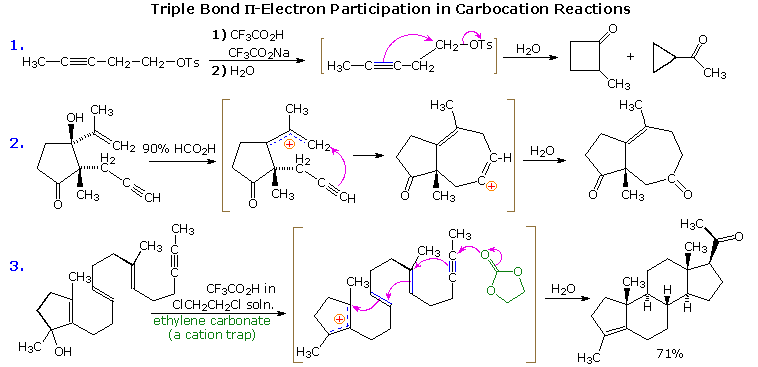 L'assistenza anchimerica tramite tripli legami non è efficace come quella tramite doppi legami, in parte perché i cationi vinilici non sono molto stabili. Nonostante ciò, i tripli legami vicinali possono catturare specie carbocationiche se acqua o altri nucleofili sono disponibili per intrappolare il risultante catione vinilico.
La reazione (1) mostra un tale esempio. L'acido trifluoroacetico è usato come solvente perché l'anione trifluoroacetato è meno nucleofilo dello ione acetato. Rese povere di questi prodotti sono trovate quando l'acido acetico è il solvente.
La reazione (2) mostra un interessante esempio di ciclizzazione iniziata dall'estremità di un catione allilico.
La reazione (3) estende l'applicazione di questa ciclizzazione ad un processo a tre passaggo, creando sei nuovi centri chirali e tre nuovi anelli. Proviene da uno studio approfondito di ciclizzazione di polieni fatto da William S. Johnson. L'acqua distrugge il catione etilene carbonato intrappolato.
L'assistenza anchimerica tramite tripli legami non è efficace come quella tramite doppi legami, in parte perché i cationi vinilici non sono molto stabili. Nonostante ciò, i tripli legami vicinali possono catturare specie carbocationiche se acqua o altri nucleofili sono disponibili per intrappolare il risultante catione vinilico.
La reazione (1) mostra un tale esempio. L'acido trifluoroacetico è usato come solvente perché l'anione trifluoroacetato è meno nucleofilo dello ione acetato. Rese povere di questi prodotti sono trovate quando l'acido acetico è il solvente.
La reazione (2) mostra un interessante esempio di ciclizzazione iniziata dall'estremità di un catione allilico.
La reazione (3) estende l'applicazione di questa ciclizzazione ad un processo a tre passaggo, creando sei nuovi centri chirali e tre nuovi anelli. Proviene da uno studio approfondito di ciclizzazione di polieni fatto da William S. Johnson. L'acqua distrugge il catione etilene carbonato intrappolato.
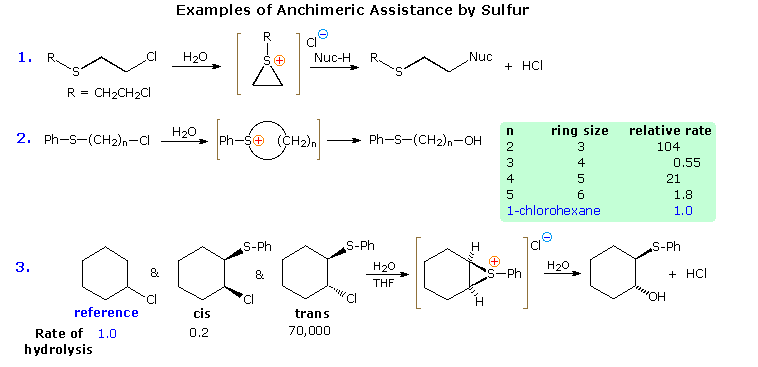 La reazione (1) è un esempio di un 'composto mostarda'. Il dicloro composto simmetrico designato da R è tristemente noto come gas mostarda, e fu usato come agente chimico durante la Prima Guerra Mondiale. Tali composti sono potenti agenti alchilanti, e reagiscono con/danneggiano una varietà di nucleofili biologici tramite cross-linking.
La reazione (2) mostra come l'influenza accelerante dello zolfo cambi all'aumentare della distanza dal gruppo uscente. L'eccezionale facilità con cui gli intermedi ciclici a tre termini sono formati riflette il piccolo angolo di legame preferito dallo zolfo divalente (ca. 90°).
La reazione (3) conferma la richiesta stereoelettronica di una relazione trans-anti dell'atomo di zolfo rispetto al gruppo uscente. Lo zolfo è molto più elettronegativo dell'idrogeno, così il suo effetto induttivo rende il cis-clorosolfuro meno reattivo del cicloesile cloruro.
La reazione (1) è un esempio di un 'composto mostarda'. Il dicloro composto simmetrico designato da R è tristemente noto come gas mostarda, e fu usato come agente chimico durante la Prima Guerra Mondiale. Tali composti sono potenti agenti alchilanti, e reagiscono con/danneggiano una varietà di nucleofili biologici tramite cross-linking.
La reazione (2) mostra come l'influenza accelerante dello zolfo cambi all'aumentare della distanza dal gruppo uscente. L'eccezionale facilità con cui gli intermedi ciclici a tre termini sono formati riflette il piccolo angolo di legame preferito dallo zolfo divalente (ca. 90°).
La reazione (3) conferma la richiesta stereoelettronica di una relazione trans-anti dell'atomo di zolfo rispetto al gruppo uscente. Lo zolfo è molto più elettronegativo dell'idrogeno, così il suo effetto induttivo rende il cis-clorosolfuro meno reattivo del cicloesile cloruro.
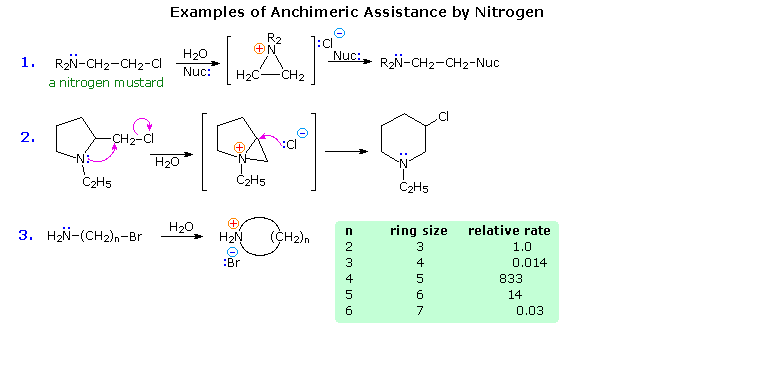 La reazione (1) illustra una classe di composti chiamati 'mostarde azotate'. Il carattere alchilante di questi composti è simile a quello dei loro analoghi solforati.
La reazione (2) mostra un caso simile in cui avviene espansione d'anello.
La reazione (3) mostra le velocità di ciclizzazione di una serie di omega-bromo alchil amine. In assenza di base, si formano sali di ammonio ciclici stabili. Le basi convertono questi sali nelle loro corrispondenti amine cicliche secondarie.
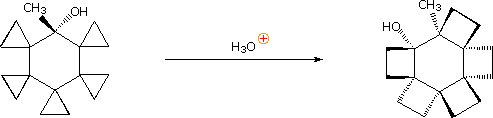
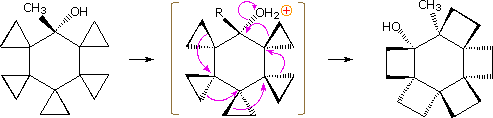
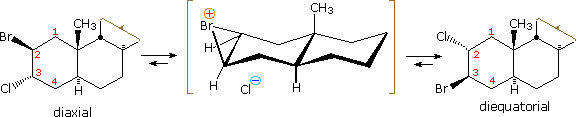
Nella maggior parte dei casi che coinvolgono l'assistenza di un eteroatomo, un intermedio "onio" viene formato, in cui l'eteroatomo è carico. Gli atomi di alogeno adicenti possono anche stabilizzare i carbocationi, come notatao in precedenza nel caso di addizioni trans-anti ad alcheni ciclici. Il riarrangiamento funzionale tramite intermedi alogenonio è stato anche riportato. Per esempio, una soluzione in cloroformio del diassiale 2-bromo-3-clorosteroide, mostrato sotto a sinistra, spontaneamente riarrangia al più stabile 2-cloro-3-bromo isomero disegnato sulla destra. Il riarrangiamento è reversibile e procede tramite ione bromonio ciclico come scritto tra parentesi.
La reazione (1) illustra una classe di composti chiamati 'mostarde azotate'. Il carattere alchilante di questi composti è simile a quello dei loro analoghi solforati.
La reazione (2) mostra un caso simile in cui avviene espansione d'anello.
La reazione (3) mostra le velocità di ciclizzazione di una serie di omega-bromo alchil amine. In assenza di base, si formano sali di ammonio ciclici stabili. Le basi convertono questi sali nelle loro corrispondenti amine cicliche secondarie.
Nella maggior parte dei casi che coinvolgono l'assistenza di un eteroatomo, un intermedio "onio" viene formato, in cui l'eteroatomo è carico. Gli atomi di alogeno adicenti possono anche stabilizzare i carbocationi, come notatao in precedenza nel caso di addizioni trans-anti ad alcheni ciclici. Il riarrangiamento funzionale tramite intermedi alogenonio è stato anche riportato. Per esempio, una soluzione in cloroformio del diassiale 2-bromo-3-clorosteroide, mostrato sotto a sinistra, spontaneamente riarrangia al più stabile 2-cloro-3-bromo isomero disegnato sulla destra. Il riarrangiamento è reversibile e procede tramite ione bromonio ciclico come scritto tra parentesi.


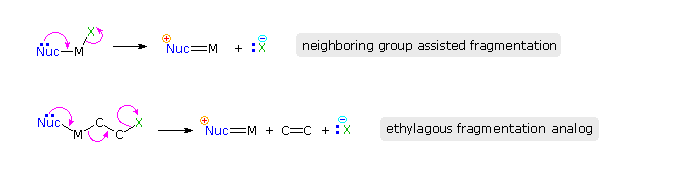
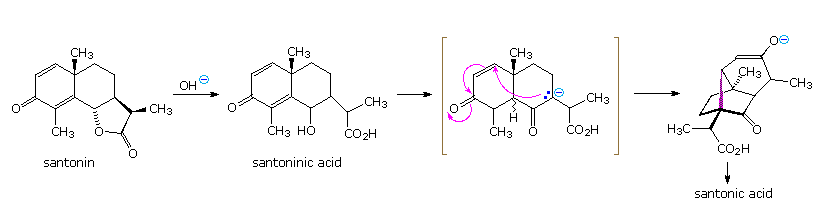
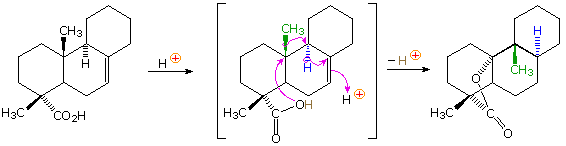
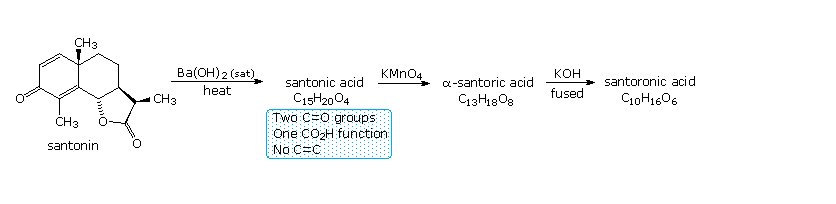 Dalle formule mostrate sopra, questa reazione non solo addizione un equivalente di acqua (prevista se il lattone viene aperto), ma crea anche un nuovo gruppo carbonile e rimuove entrambi i doppi legami carbonio-carbonio. La natura della trasformazione della santonina ad acido santonico rimase sconosciuta per molti anni, ma fu risolta nel 1948 grazie al lavoro di R. B. Woodward e collaboratori ad Harvard.
Nello schema sotto sono mostrate le strutture dell'acido santonico e dei suoi prodotti ossidativi.
Dalle formule mostrate sopra, questa reazione non solo addizione un equivalente di acqua (prevista se il lattone viene aperto), ma crea anche un nuovo gruppo carbonile e rimuove entrambi i doppi legami carbonio-carbonio. La natura della trasformazione della santonina ad acido santonico rimase sconosciuta per molti anni, ma fu risolta nel 1948 grazie al lavoro di R. B. Woodward e collaboratori ad Harvard.
Nello schema sotto sono mostrate le strutture dell'acido santonico e dei suoi prodotti ossidativi.
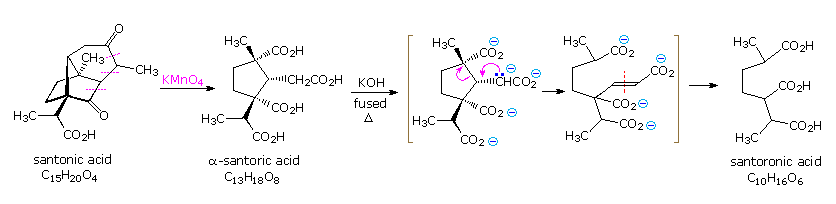 Il meccanismo attraverso cui questo cambiamente degno di nota avviene è mostrato sotto. L'iniziale idrolisi del lattone ad acido santoninico è seguita da isomerismo dell'insaturazione α,β a β,ν e tautomerizzazione. Il risultante dichetone, nella sua configurazione cis-fusa, subisce quindi una reazione di Michael intramolecolare, formando un nuovo anello a cinque termini (notare il legame colorato).
Il meccanismo attraverso cui questo cambiamente degno di nota avviene è mostrato sotto. L'iniziale idrolisi del lattone ad acido santoninico è seguita da isomerismo dell'insaturazione α,β a β,ν e tautomerizzazione. Il risultante dichetone, nella sua configurazione cis-fusa, subisce quindi una reazione di Michael intramolecolare, formando un nuovo anello a cinque termini (notare il legame colorato).