Geber
2021-03-19 12:33
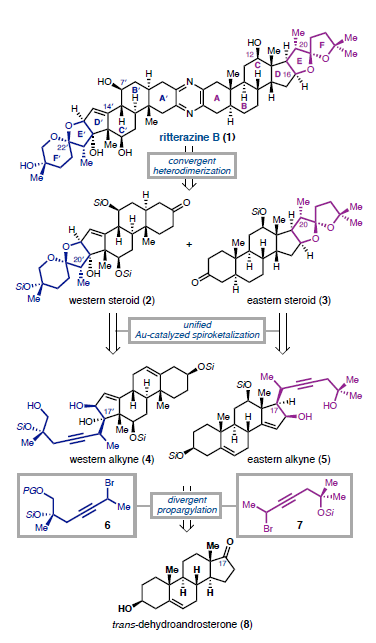
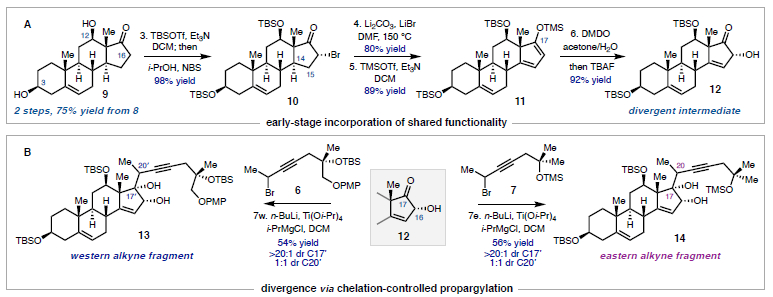
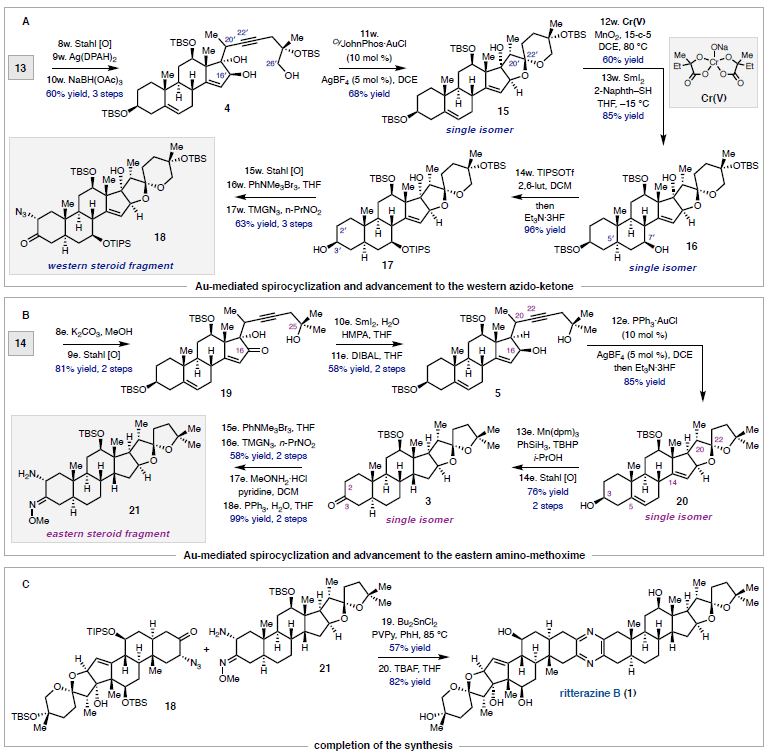
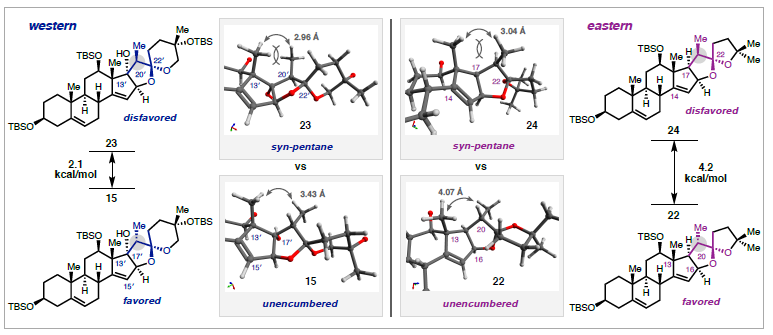
La ritterazina B (vedi immagine sotto) è un prodotto naturale a struttura pirazinica bis-steroidea (BSP) che fu isolato nel 1995 dal tunicate marino Riterella tokioka al largo della penisola del Giappone.1 I BSP includono alcuni dei più potenti composti anticancro scoperti finora,2 e la ritterazina B in particolare è stata descritta come “tra i più potenti inibitori della crescita mai testati” dal National Cancer Institute (NCI).3-5 Essa possiede attività sub-nanomolare contro le cellule della leucemia P388 (0.17 nM GI50)6 ed GI50 medio di 3.2 nM nello screening della linea cellulare NCI-60.5,7 Dato che i BPS mostrano distinti modelli di attività nelle analisi NCI-60 COMPARE, è stato proposto che essi agiscano tramite un distinto modo di azione rispetto alle chemoterapie esistenti.4,7,8 Sebbene i BSP siano noti nell'indurre apoptosi,3 una mancanza di materiale naturale ha ostacolato le indagini traduzionali della ritterazina B e relativi composti. Studi di riferimento fatti da Shair e collaboratori implicavano i BSP come leganti ad alta affinità per proteine che legano l'ossisterolo,4 mentre prove più recenti hanno suggerito che la proteina sensibile al calore specifica del reticolo endoplasmatico GRP78 può essere il loro target efficace.9,10 Dati questi promettenti studi fondamentali, è necessaria una via sintetica migliorata per ottenere la ritterazina B per valutare completamente il suo potenziale come chemioterapico.2,8 In questa comunicazione, viene riportata la prima sintesi totale della ritterazina B. L'approccio usa una strategia comune per preparare entrambi gli spirochetali steroidei dal trans-deidroandrosterone, uno steroide commercialmente disponibile e non costoso. In linea con i procedenti tentativi di accedere ai BSP,5,11 l'analisi retrosintetica è iniziata con la scissione dell'anello pirazinico centrale, rivelando gli steroidi western e eastern 2 e 3, rispettivamente. Per semplificare lo sviluppo del percorso, si è cercato di preparare entrambi 2 e 3 da un materiale di partenza comune, utilizzando la stessa tattica generale per la formazione di legami C-C e per la spirochetalizzazione. In questo senso, gli steroidi 2 e 3 sono stati semplificati nei corrispondenti alchini 4 e 5, dove la cicloisomerizzazione12 catalizzata da metalli di transizione verrebbe usata per formare i rispettivi spirochetali. Questo passaggio retrosintetico ha spostato la sfida sintetica sull'unione di frammenti alchinici differenziati con un comune centro steroideo. Si è immaginato di preparare gli alchini 4 e 5 tramite addizione 1,2 della specie propargile-metallo derivata da 6 o 7 ad un α-idrossi chetone accessibile dal trans-deidroandrosterone (8). La scelta di 8 quale materiale di partenza è stata vista come strategica: l'alchene C5–C6 fornirebbe un appiglio per l'ossidazione dell'anello B in uno stadio avanzato. Questa tattica è già stata usata in approcci sintetici ai BPS,11 che potrebbe essere il motivo per cui i BSP con ossidazione C7/C7ʹ non sono stati precedentemente sintetizzati.7 Andando avanti, il noto steroide 9 (preparato in due passaggi da 8)13 è stato trattato con un eccesso di tert-butildimetilsililtriflato e trietilammina (Et3N) per proteggere gli alcoli al C3 ed al C12 e formare un silil enol etere al C17 (Schema 1A). La addizione diretta di isopropanolo ed N-bromosuccinimmide alla miscela di reazione ha prodotto l'α-bromo-chetone 10 con una resa quantitativa in un solo passaggio. L'eliminazione del bromuro al C16 in condizioni basiche ha fornito una miscela insignificante di enoni isomerici (Δ14–15 e Δ15–16, non mostrati), che convergevano al dienol etere 11 per trattamento con Et3N e trimetilsilil triflato. L'epossidazione selettiva dell'alchene C16–C17 con dimetildiossirano e la successiva addizione di tetrabutilammonio fluoruro (TBAF) ha fornito l'α-idrossi-chetone 12 con una resa de 92%, che servirebbe come intermedio divergente. A questo punto, rivogliamo la nostra attenzione nel preparare i distinti spirochetali trovati negli steroidi western ed eastern 2 e 3, rispettivamente. A tal fine, le propargilazioni mediate da titanio basate sulle condizioni riportate da Sato e collaboratori si sono dimostrate particolarmente efficaci (Schema 1B).14 La deprotonazione dell'alcol al C16 attraverso trattamento del 12 con n-butillitio, seguita dall'aggiunta delle specie organotitanio derivate da entrambi i propargil bromuri 6 o 7 hanno portato all'addizione 1,2 fornendo l'alchino 13 con una resa del 54% o l'alchino 14 con una resa del 56%. Queste addizioni sono avvenute con una selettività esclusiva per la faccia β nonostante il gruppo metile assiale,15 forse a causa della formazione di un chelato ciclico α-eliminato tra gli ossigeni al C16 e al C17. Mentre è stata ottenuta una eccellente diastereoselettività al C17, 13 e 14 si sono formati come miscele 1:1 di epimeri al C20ʹ/C20. Mentre l'α-stereoisomere dell'alcol al C16 è stato cruciale per impartire lo stereocontrollo desiderato nelle reazioni di propargilazione, erano richiesti alcoli β-orientati per elaborare gli spirochetali richiesti. Per la preparazione dello steroide western (2), la stereoinversione è stata ottenuto tramite ossidazione nell'enone seguita da scissione del p-metossifenil etere16 e riduzione 1,2 diretta dall'idrossile, che ha fornito lo spirochetale precursore 4 con una resa dell'86% (Schema 2A). Dopo estesa sperimentazione,17 il trattamento del diolo 4 con CyJohnPhos·AuCl (10 mol %) catalitico e AgBF4 (5 mol %) ha fornito lo spirochetale 15 con una resa del 68% come singolo diastereoisomero.12 Per la nostra gioia, questa reazione non solo ha fornito la corretta configurazione sullo spirochetale, ma è anche proceduta con convergenza degli epimeri al C20ʹ, fornendo il composto 15 con la α-disposizione richiesta del gruppo metile. La selettività totale è stato trovato essere criticamente dipendente dalla scelta del solvente diclorometano, dal ligando CyJohnPhos, e dal controione tetrafluoroborato. Con lo spirociclo western in mano, l'ossidazione allilica del 15 al C7ʹ è stata realizzata con l'ossocromato (Cr(V)).18 Per ottenere l'anello Bʹ completamente saturo, l'enone intermedio (non mostrato) è stato ridotto con SmI2 e 2-naftalene tiolo per fornire 16 con una resa dell'85% come singolo diastereoisomero; le basse temperature sono state cruciali per prevenire la sovraossidazione dell'alchene a C14–C15.17 La protezione dell'alcol al C7ʹ di 16 come tri-isopropilsilil etere è stata seguita dall'aggiunta di Et3N·3HF per rivelare selettivamente l'alcol al C3ʹ nello stesso pallone di reazione. L'ossidazione di 17 a chetone e l'α-bromurazione e l'azidazione in due passaggi al C2 usando le procedure sviluppate da Shair11i e Fuchs11d ha fornito il frammento western come cheto-azide 18 con una resa totale del 63%. L'uso del nitropropano come solvente per l'azidazione invece del tradizionale nitrometano è stata scoperta necessaria per la solubilizzazione dell'intermedio bromuro.11d,i Questo ha anche aumentato significativamente la resa prevenendo l'indesiderata eliminazione di N2 da 18. La sintesi dello steroide eastern ha seguito una sequenza simile a quella descritta sopra, ma è stata modificata leggermente per preparare il diolo 5 (Schema 2B). Seguendo la deprotezione del trimetilsilil etere in 14 e l'ossidazione di Stahl, l'alcol al C17 è stato rimosso via deossigenazione del chetolo con SmI2 e H2O.19 La riduzione 1,2 diastereoselettiva è stata effettuata tramite il trattamento con di-iso-butilalluminio idruro a dare 5; la riduzione a quattro elettroni di 19 direttamente a dare 5 potrebbe essere ottenuta con un eccesso di SmI2, sebbene la resa e la diastereoselettività è stata fortemente ridotta rispetto alla procedura in due passaggi. Come osservato per il frammento western, la spirociclizzazione catalizzata da Au(I) è proceduta senza problemi nel costruire il sistema ciclico 5/5 (E/F), ancora come singolo isomero sia al C20 sia al C22. Qui, è statp scoperto che l'addizione diretta di Et3N·3HF alla miscela di reazione ha portato alla deprotezione selettiva del silil etere al C3, fornendo in ultima analisi 20 con una resa dell'85%. I due stereocentri rimanenti richiesti per la il frammento eastern sono stati installati in un singolo passaggio tramite riduzione per transfer di atomo di idrogeno (HAT) degli alcheni a C5–C6 e C14–C15 nelle condizioni sviluppate da Shenvi e collaboratori.20 Il prodotto completamente saturo è stato ottenuto con una fusione cis alla giunzione degli anelli C/D; studi DFT supportano la preferenza termodinamica per la stereochimica osservata.17,20 Questa reazione si è dimostrata critica per l'accesso al materiale in stadio avanzato nel corretto stato di ossidazione, dato che le tipiche condizioni di idrogenazione di un alchene sono state incapaci di ridurre il doppio legame al C14–C15 double bond. Per completare il partner di accoppiamento eastern, è stato seguito il medesimo protocollo di ossidazione/bromurazione/azidazione come descritto per il frammento western tramite condensazione del chetone con MeONH2·HCl e riduzione di Staudinger21 per fornire l'ammino-metossima 21 con una resa quantitativa.11d L'eterodimerizzazione nella condizioni di catalisi acida di Lewis, come riportato originariamente da Fuchs,11d hanno formito la piraziona desiderata (Schema 2C). La deprotezione globale con TBAF11i ha consegnato la ritterazina B con una resa dell'82%, rappresentando la sua prima sintesi totale. I dati di caratterizzazione spettroscopica collimavano con quelli riportati per il materiale naturale.1 La costruzione di ogni frammento è imperniato sulla reazione modulare di spirochetalizzazione. Data l'epimerizzazione osservata a C20/C20ʹ, si è ipotizzato che il processo avvenga tramite una iniziale monociclizzazione/isomerizzazione stereoablativa, seguita da una formazione del chetale doppiamente diastereoselettiva.5,12a Per sondare la formazione preferenziale degli stereocentri a C20/C20ʹ, le energie dello stato fondamentale sono state calcolare per i composti 15 e 22 ottimizzati tramite DFT. Inoltre, gli isomeri osservati sono stati scoperti essere di 2.1 e 4.2 kcal/mol inferiori in energia rispetto agli epimeri innaturali al C20 e al C20ʹ, rispettivamente.22 L'analisi conformazionale delle specie sfavorite 23 e 24 ha rilevato l'esistenza di interazioni del tipo syn pentano tra i β-Me a C20/C20ʹ e i gruppi Me assiali a C13/C13ʹ.5,23 Queste interazioni non sono presenti con i gruppi Me disposti α al C20, che appaiono meno stericamente ingombrati. Riassumendo, la prima sintesi totale della ritterazina B è stata completata partendo dal semplice steroide trans-deidroandrosterone ed usando un approccio unificato ad entrambi i frammenti steroidei. Le caratteristiche chiave della strategia includono le propargilazioni mediate da titanio per accedere ad alchini differenziati così come le spirociclizzazioni oro-catalizzate, diastereoselettive. La modularità di questo approccio ha permesso l'accesso a tre addizionali BSP finora, inclusi due nuovi composti non naturali. Indagini sull'attività biologica della ritterazina B e relativi composti sono attualmente in corso. Vale la pena notare che sono stati preparati finora diversi lotti multi-milligrammo di questi materiali, e anche se si è scelto di eseguire i passaggi finali su piccola scala per sicurezza, la sintesi dei frammenti da accoppiare si è rivelata essere scalabile. Ci si aspetta che questa via sintetica sviluppata oggi fornirà ampio materiale per studi biologici, permettendo ulteriori indagini dei BSP come terapeutici antitumorali. REFERIMENTI (1) Fukuzawa, S.; Matsunaga, S.; Fusetani, N. Isolation and Structure Elucidation of Ritterazines B and C, Highly Cytotoxic Dimeric Steroidal Alkaloids, from the Tunicate Ritterella Tokioka. J. Org. Chem. 1995,60 (3), 608–614. (2) Moser, B. R. Review of Cytotoxic Cephalostatins and Ritterazines: Isolation and Synthesis. J. Nat. Prod. 2008, 71 (3), 487–491. (3) Komiya, T.; Fusetani, N.; Matsunaga, S.; Kubo, A.; Kaye, F. J.; Kelley, M. J.; Tamura, K.; Yoshida, M.; Fukuoka, M.; Nakagawa, K. Ritterazine B, a New Cytotoxic Natural Compound, Induces Apoptosis in Cancer Cells. Cancer Chemother. Pharmacol. 2003, 51(3), 202–208. (4) Burgett, A. W. G.; Poulsen, T. B.; Wangkanont, K.; Anderson, D. R.; Kikuchi, C.; Shimada, K.; Okubo, S.; Fortner, K. C.; Mimaki, Y.; Kuroda, M.; Murphy, J. P.; Schwalb, D. J.; Petrella, E. C.; Cornella-Taracido, I.; Schirle, M.; Tallarico, J. A.; Shair, M. D. Natural Products Reveal Cancer Cell Dependence on Oxysterol-Binding Proteins. Nat. Chem. Biol. 2011, 7 (9), 639–647. (5) Phillips, S. T.; Shair, M. D. Syntheses of the Eastern Halves of Ritterazines B, F, G, and H, Leading to Reassignment of the 5,5- Spiroketal Stereochemistry of Ritterazines B and F. J. Am. Chem. Soc. 2007, 129 (20), 6589–6598. (6) Fukuzawa, S.; Matsunaga, S.; Fusetani, N. Isolation of 13 New Ritterazines from the Tunicate Ritterella Tokioka and Chemical Transformation of Ritterazine B1. J. Org. Chem. 1997, 62 (13), 4484–4491. (7) Budzikiewicz, H.; Flessner, T.; Jautelat, R.; Scholz, U.; Winterfeldt, E. Progress in the Chemistry of Organic Natural Products; Springer Vienna: Vienna, 2004. (8) Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M. Alkaloids from Marine Invertebrates as Important Leads for Anticancer Drugs Discovery and Development. Molecules 2014, 19 (12), 20391–20423. (9) López-Antón, N.; Rudy, A.; Barth, N.; Schmitz, L. M.; Pettit, G. R.; Schulze-Osthoff, K.; Dirsch, V. M.; Vollmar, A. M. The Marine Product Cephalostatin 1 Activates an Endoplasmic Reticulum Stress-Specific and Apoptosome-Independent Apoptotic Signaling Pathway. J. Biol. Chem. 2006, 281 (44), 33078–33086. (10) Ambrose, A. J.; Santos, E. A.; Jimenez, P. C.; Rocha, D. D.; Wilke, D. V.; Beuzer, P.; Axelrod, J.; Kumar Kanduluru, A.; Fuchs, P. L.; Cang, H.; Costa-Lotufo, L. V.; Chapman, E.; La Clair, J. J. Ritterostatin GN1N, a Cephalostatin–Ritterazine Bis-Steroidal Pyrazine Hybrid, Selectively Targets GRP78. ChemBioChem 2017, 18 (6), 506–510. (11) (a) Jeong, J. U.; Fuchs, P. L. Synthesis of a 17-Deoxy, C-14,15-Dihydro Derivative of the North Spiroketal Moiety of the Cephalostatins. Conversion to a (+)-Trisdecacyclic C2 Symmetrical Pyrazine. J. Am. Chem. Soc. 1994, 116 (2), 773–774. (b) Heathcock, C. H.; Smith, S. C. Synthesis and Biological Activity of Unsymmetrical Bis-Steroidal Pyrazines Related to the Cytotoxic Marine Natural Product Cephalostatin 1. J. Org. Chem. 1994, 59 (22), 6828–6839. (c) Jeong, J. U.; Sutton, S. C.; Kim, S.; Fuchs, P. L. Biomimetic Total Syntheses of (+)-Cephalostatin 7, (+)-Cephalostatin 12, and (+)-Ritterazine K. J. Am. Chem. Soc. 1995, 117 (40), 10157–10158. (d) Guo, C.; Bhandaru, S.; Fuchs, P. L.; Boyd, M. R. An Efficient Protocol for the Synthesis of Unsymmetrical Pyrazines. Total Synthesis of Dihydrocephalostatin 11. J. Am. Chem. Soc. 1996, 118 (43), 10672–10673. (e) LaCour, T. G.; Guo, C.; Bhandaru, S.; Boyd, M. R.; Fuchs, P. L. Interphylal Product Splicing: The First Total Syntheses of Cephalostatin 1, the North Hemisphere of Ritterazine G, and the Highly Active Hybrid Analogue, Ritterostatin GN1N1. J. Am. Chem. Soc. 1998, 120 (4), 692–707. (f) Jeong, J. U.; Guo, C.; Fuchs, P. L. Synthesis of the South Unit of Cephalostatin. 7. Total Syntheses of (+)-Cephalostatin 7, (+)-Cephalostatin 12, and (+)-Ritterazine K1. J. Am. Chem. Soc. 1999, 121 (10), 2071–2084. (g) Kim, S.; Sutton, S. C.; Guo, C.; LaCour, T. G.; Fuchs, P. L. Synthesis of the North 1 Unit of the Cephalostatin Family from Hecogenin Acetate1. J. Am. Chem. Soc. 1999, 121 (10), 2056–2070. (h) Lee, S.; Fuchs, P. L. The First Total Synthesis of (Corrected) Ritterazine M. Org. Lett. 2002, 4 (3), 317–318. (i) Fortner, K. C.; Kato, D.; Tanaka, Y.; Shair, M. D. Enantioselective Synthesis of (+)-Cephalostatin 1. J. Am. Chem. Soc. 2010, 132 (1), 275–280. (12) (a) Liu, B.; De Brabander, J. K. Metal-Catalyzed Regioselective Oxy-Functionalization of Internal Alkynes: An Entry into Ketones, Acetals, and Spiroketals. Organic Letters 2006, 8 (21), 4907–4910. (b) Tlais, S. F.; Dudley, G. B. A Gold-Catalyzed Alkyne-Diol Cycloisomerization for the Synthesis of Oxygenated 5,5-Spiroketals. Beilstein J. Org. Chem. 2011, 7, 570–577. (c) Goodwin, J. A.; Aponick, A. Regioselectivity in the Au-Catalyzed Hydration and Hydroalkoxylation of N-Alkynes. Chem. Commun. 2015, 51 (42), 8730–8741. (d) Pflästerer, D.; Rudolph, M.; Hashmi, A. S. K. Gold-Catalyzed Hydrofunctionalizations and Spiroketalizations of Alkynes as Key Steps in Total Synthesis. Isr. J. Chem. 2018, 58 (5), 622–638. (13) (a) Schönecker, B.; Lange, C.; Zheldakova, T.; Günther, W.; Görls,H.; Vaughan, G. Copper-Mediated Regio- and Stereoselective 12β- Hydroxylation of Steroids with Molecular Oxygen and an Unexpected 12β-Chlorination. Tetrahedron 2005, 61 (1), 103–114. (b) Trammell, R.; See, Y. Y.; Herrmann, A. T.; Xie, N.; Díaz, D. E.; Siegler, Maxime. A.; Baran, P. S.; Garcia-Bosch, I. Decoding the Mechanism of Intramolecular Cu-Directed Hydroxylation of sp3 C–H Bonds. J. Org. Chem. 2017, 82 (15), 7887–7904. (14) Nakagawa, T.; Kasatkin, A.; Sato, F. Highly Efficient Synthesis of Propargyl- and Allenyltitanium Reagents from Propargyl Halides or Propargyl Alcohol Derivatives. Practical Synthesis of Allenyl and Homopropargyl Alcohols. Tetrahedron Lett. 1995, 36 (18), 3207–3210. (15) (a) Smith, H.; Huges, G. A.; Douglas, G. H.; Wendt, G. R.; Buzby Jr., G. C.; Edgren, R. A.; Fisher, J.; Foell, T.; Gadsby, B.; Hartley, D.; Herbst, D.; Jansen, A. B. A.; Ledig, K.; McLoughlin, B. J.; McMenamin, J.; Pattison, T. W.; Phillips, P. C.; Rees, R.; Siddall, J.; Siuda, J.; Smith, L. L.; Tokolics, J.; Watson, D. H. P. Totally Synthetic Steroid Hormones. Part II. 13b-Alkylgona-1,3,5(10)-Trienes, 13b-Alkygon-4-En-3-Ones, and Related Compounds. J. Chem. Soc. 1964, 4472–4492. (b) McKinney, A. R.; Suann, C. J.; Stenhouse, A. M. A Stereochemical Examination of the Equine Metabolism of 17α-Methyltestosterone. Anal. Chim. Acta 2007, 581 (2), 377–387. (16) Noshita, T.; Sugiyama, T.; Kitazumi, Y.; Oritani, T. Phenolic Ferrier Reaction and Its Application to the Natural Product Synthesis. Tetrahedron Lett. 1994, 35 (44), 8259–8262. (17) See the Supporting Information. (18) (a) Krumpolc, M.; Rocek, J. Chromium(V) Oxidations of Organic Compounds. Inorg. Chem. 1985, 24 (4), 617–621. (b) Kanda, Y.; Ishihara, Y.; Wilde, N. C.; Baran, P. S. Two-Phase Total Synthesis of Taxanes: Tactics and Strategies. J. Org. Chem. 2020, 85 (16), 10293– 10320. (19) Szostak, M.; Spain, M.; Procter, D. J. Recent Advances in the Chemoselective Reduction of Functional Groups Mediated by Samarium(II) Iodide: A Single Electron Transfer Approach. Chem. Soc. Rev. 2013, 42 (23), 9155–9183. (20) Iwasaki, K.; Wan, K. K.; Oppedisano, A.; Crossley, S. W. M.; Shenvi, R. A. Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc. 2014, 136 (4), 1300–1303. (21) Staudinger, H.; Meyer, J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 1919, 2 (1), 635–646. (22) Calcolato usando il pacchetto software entos al livello teorico BLYP/6-31G**. See Manby, F.; Miller, T.; Bygrave, P.; Ding, F.; Dresselhaus, T.; Batista-Romero, F.; Buccheri, A.; Bungey, C.; Lee, S.; Meli, R.; Miyamoto, K.; Steinmann, C.; Tsuchiya, T.; Welborn, M.; Wiles, T.; Williams, Z. Entos: A Quantum Molecular Simulation Package. ChemRxiv 2019, No. 7762646.v2. (23) Chen, R.; Shen, Y.; Yang, S.; Zhang, Y. Conformational Design Principles in Total Synthesis. Angew. Chem., Int. Ed. 2020, 59 (34), 14198–14210.
I seguenti utenti ringraziano Geber per questo messaggio: EdoB