quimico
2011-11-24 15:12
Introduzione e background
Le alghe rosse e gli organismi marini che si nutrono di alghe rosse, in particolare della specie Laurencia, hanno prodotto diverse acetogenine C15 non terpenoidi contenenti eteri ciclici di medie dimensioni alogenati1. Questi metaboliti contengono un numero di diversi dimensioni degli anelli, come quelle trovate nella (+)-laurencina2, (+)-prelaureatina3, (+)-laurallene4, (-)-isolaurallene5 e (+)-ottusenina6 (come mostrato sotto).
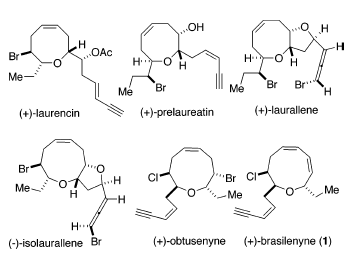 Tra esse, la (+)-brasilenina (1), isolata dalla ghiandola digestiva dell'Aplysia brasiliana da Fenical et al.7 nel 1979, ha un nuovo scheletro etereo ciclico a nove membri contenente un'unità dienica
1,3-cis,cis. Poiché tali organismi sono incapaci di manovre evasive, mancando predatori significativi, suggerisce che i metaboliti secondari, come 1, siano prodotti e/o concentrati nella ghiandola digestiva come sostanze chimiche di difesa.
Quindi, gli studi in vivo di 1 e della (+)-cis-diidrorodofitina, un componente maggioritario dalla stessa fonte naturale, avevano quindi dimostrato che questi erano potenti inibitori dei normali comportamenti nutritivi7a.
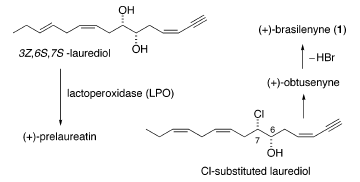
Indagini biosintetiche sui metaboliti eterei ciclici della specie Laurencia hanno dimostrato che la lattoperossidasi (LPO) trasforma direttamente il 3Z,6S,7S-laurediolo, che si trova in natura come diversi stereoisomeri, in (+)-prelaureatina attraverso formazioni di etere indotta dallo ione bromonio8. Inoltre, (+)-laurallene, (+)-laureatina, e (+)-isolaureatina si presume derivino da (+)-prelaureatina tramite un simile cammino biogenetico3,9. Perciò, l'intermedio ipotetico, un laurediolo Cl sostituito, è stato postulato produrre (+)-ottusenina tramite una simile biotransformazione, e la successiva deidrobromurazione
porta all'ottenimento della (+)-brasilenina7.
L'ipotesi è stata supportata dalla scoperta che quei composti hanno la stessa configurazione al C(6) e C(7) dell'intermedio Cl sostituito10 (schema sottostante).
Tra esse, la (+)-brasilenina (1), isolata dalla ghiandola digestiva dell'Aplysia brasiliana da Fenical et al.7 nel 1979, ha un nuovo scheletro etereo ciclico a nove membri contenente un'unità dienica
1,3-cis,cis. Poiché tali organismi sono incapaci di manovre evasive, mancando predatori significativi, suggerisce che i metaboliti secondari, come 1, siano prodotti e/o concentrati nella ghiandola digestiva come sostanze chimiche di difesa.
Quindi, gli studi in vivo di 1 e della (+)-cis-diidrorodofitina, un componente maggioritario dalla stessa fonte naturale, avevano quindi dimostrato che questi erano potenti inibitori dei normali comportamenti nutritivi7a.
Indagini biosintetiche sui metaboliti eterei ciclici della specie Laurencia hanno dimostrato che la lattoperossidasi (LPO) trasforma direttamente il 3Z,6S,7S-laurediolo, che si trova in natura come diversi stereoisomeri, in (+)-prelaureatina attraverso formazioni di etere indotta dallo ione bromonio8. Inoltre, (+)-laurallene, (+)-laureatina, e (+)-isolaureatina si presume derivino da (+)-prelaureatina tramite un simile cammino biogenetico3,9. Perciò, l'intermedio ipotetico, un laurediolo Cl sostituito, è stato postulato produrre (+)-ottusenina tramite una simile biotransformazione, e la successiva deidrobromurazione
porta all'ottenimento della (+)-brasilenina7.
L'ipotesi è stata supportata dalla scoperta che quei composti hanno la stessa configurazione al C(6) e C(7) dell'intermedio Cl sostituito10 (schema sottostante).
 L'interessante struttura di questi metaboliti marini ha stimolato un significativo livello di impegno per la costruzione dei sistemi ad anello ossonina ed ossocene11. Recenti esempi rappresentativi inclusono la sintesi di Crimmins’ della (+)-prelaureatina11b e della (+)-ottusenina11e attraverso aldolica o una sequenza di alchilazione/metatesi di chiusura d'anello (RCM), la sintesi di Overman della (-)-laurenina11j e della (+)-laurencina11k attraverso ciclizzazione alchene-acetale promossa da acido di Lewis, e la sintesi di Holmes della (+)-laurencina11l che sfrutta un riarrangiamento di Claisen. Questi metodi sono particolarmente adatti per la costruzione di eteri di medie dimensioni d'anello contenenti un singolo doppio legame carbonio-carbonio. La sfida sintetica costituita dal nucleo ossoninico di 1, comunque, richiede la formazione di un anello di medie dimensioni che porta un diene coniugato. Due recenti report (Negishi et al.12 e Isobe et al.13) descrivono metodi percorribili per la sintesi di anelli di medie dimensioni che contengono un 1,3-diene. Questi report contengono la carbopalladazione
di un allene e la ciclizzazzione acido catalizzata di un complesso acetilene dicobalto.
Quale parte del programma degli autori sullo sviluppo di nuove reazioni di cross-coupling basate su silicio, essi hanno dimostrato in precedenza il potenziale sintetico della reazione sequenziale di ring-closing metathesis (RCM)/cross-coupling intramolecolare assistito da silicio per la costruzione di sistemi di medie dimensioni, carbo- ed eterociclici che recano un'unità 1,3-cis,cis-dienica14. Questo processo di coupling è idealmente adatto per generare il nucleo ossoninico di 1, con il suo 1,3-diene interno. Comunque, questo approccio introduce diverse sfide che richiedono ulteriori manipolazioni (schema sotto). In contrasto con gli studi metodologici precedenti su sistemi più semplici, la catena laterale in C(9) ed il gruppo etile in C(2) hanno presentato potenziali difficoltà per il processo di coupling intramolecolare. Inoltre, la presenza del centro recante cloro in C(8) ha richiesto la creazione di un gruppo funzionale idrossile in C(8) di configurazione opposta. Questo, a turno, permette l'uso di una catena temporanea di silicio per costruzione dell'alchenilsilil etere ciclico tramite RCM. La creazione del centro stereogenico in C(2) è l'altra parte critica della strategia necessaria per la sintesi di 1. Questo problema ha stimolato lo sviluppo di una nuova reazione di apertura d'anello dell'1,3-diossolanone con un nucleofilo acetilenico per creare il centro stereogenico richiesto in posizione propargilica.
L'interessante struttura di questi metaboliti marini ha stimolato un significativo livello di impegno per la costruzione dei sistemi ad anello ossonina ed ossocene11. Recenti esempi rappresentativi inclusono la sintesi di Crimmins’ della (+)-prelaureatina11b e della (+)-ottusenina11e attraverso aldolica o una sequenza di alchilazione/metatesi di chiusura d'anello (RCM), la sintesi di Overman della (-)-laurenina11j e della (+)-laurencina11k attraverso ciclizzazione alchene-acetale promossa da acido di Lewis, e la sintesi di Holmes della (+)-laurencina11l che sfrutta un riarrangiamento di Claisen. Questi metodi sono particolarmente adatti per la costruzione di eteri di medie dimensioni d'anello contenenti un singolo doppio legame carbonio-carbonio. La sfida sintetica costituita dal nucleo ossoninico di 1, comunque, richiede la formazione di un anello di medie dimensioni che porta un diene coniugato. Due recenti report (Negishi et al.12 e Isobe et al.13) descrivono metodi percorribili per la sintesi di anelli di medie dimensioni che contengono un 1,3-diene. Questi report contengono la carbopalladazione
di un allene e la ciclizzazzione acido catalizzata di un complesso acetilene dicobalto.
Quale parte del programma degli autori sullo sviluppo di nuove reazioni di cross-coupling basate su silicio, essi hanno dimostrato in precedenza il potenziale sintetico della reazione sequenziale di ring-closing metathesis (RCM)/cross-coupling intramolecolare assistito da silicio per la costruzione di sistemi di medie dimensioni, carbo- ed eterociclici che recano un'unità 1,3-cis,cis-dienica14. Questo processo di coupling è idealmente adatto per generare il nucleo ossoninico di 1, con il suo 1,3-diene interno. Comunque, questo approccio introduce diverse sfide che richiedono ulteriori manipolazioni (schema sotto). In contrasto con gli studi metodologici precedenti su sistemi più semplici, la catena laterale in C(9) ed il gruppo etile in C(2) hanno presentato potenziali difficoltà per il processo di coupling intramolecolare. Inoltre, la presenza del centro recante cloro in C(8) ha richiesto la creazione di un gruppo funzionale idrossile in C(8) di configurazione opposta. Questo, a turno, permette l'uso di una catena temporanea di silicio per costruzione dell'alchenilsilil etere ciclico tramite RCM. La creazione del centro stereogenico in C(2) è l'altra parte critica della strategia necessaria per la sintesi di 1. Questo problema ha stimolato lo sviluppo di una nuova reazione di apertura d'anello dell'1,3-diossolanone con un nucleofilo acetilenico per creare il centro stereogenico richiesto in posizione propargilica.
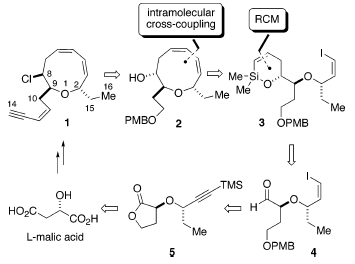 Il piano di sintesi formulato per la sintesi della (+)-brasilenina è delineato retrosinteticamente nello schema sovrastante. La semplificazione della catena laterale eninica e la funzionalità cloruro riducono la sfida costituita dall'intermedio 2, che era stato progettato derivare da una reazione di cross-coupling intramolecolare palladio-catalizzata, assistita da silicio di 3. Usando il silossano ciclico a sei membri, il gruppo idrossile liberato nel cross-coupling è perfettamente situato per l'installazione del cloruro in C(8). L'alchenilsilil etere ciclico 3 dovrebbe derivare da un'allilazione diastereoselettiva dell'aldeide 4 e dall'applicazione dell ring-closing metathesis (RCM) del derivato vinil alchenilsilil etere. L'aldeide 4 con un gruppo idrossilee primario protetto, così come il vinil ioduro geometricamente definito dovrebbe essere, senza difficoltà, è stato sviluppato da 5. La sintesi diastereo- ed enantioselettiva di 5 hanno rappresentato una sfida sintetica affascinante, cioè la costruzione di un etere doppiamente ramificato con centri stereogenici affiancati (schema sottostante).
Il piano di sintesi formulato per la sintesi della (+)-brasilenina è delineato retrosinteticamente nello schema sovrastante. La semplificazione della catena laterale eninica e la funzionalità cloruro riducono la sfida costituita dall'intermedio 2, che era stato progettato derivare da una reazione di cross-coupling intramolecolare palladio-catalizzata, assistita da silicio di 3. Usando il silossano ciclico a sei membri, il gruppo idrossile liberato nel cross-coupling è perfettamente situato per l'installazione del cloruro in C(8). L'alchenilsilil etere ciclico 3 dovrebbe derivare da un'allilazione diastereoselettiva dell'aldeide 4 e dall'applicazione dell ring-closing metathesis (RCM) del derivato vinil alchenilsilil etere. L'aldeide 4 con un gruppo idrossilee primario protetto, così come il vinil ioduro geometricamente definito dovrebbe essere, senza difficoltà, è stato sviluppato da 5. La sintesi diastereo- ed enantioselettiva di 5 hanno rappresentato una sfida sintetica affascinante, cioè la costruzione di un etere doppiamente ramificato con centri stereogenici affiancati (schema sottostante).
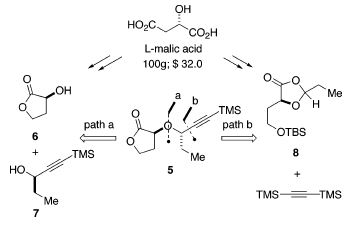 La soluzione seemplice a questo problema dovrebbe ricorrere ad una reazione di sostituzione nucleofila. Poiché entrambi gli enantiomeri di 615 e 716 sono disponibili, l'unione dell'etere doppiamente ramificato può procedere in entrambe le direzioni. Questo approccio era considerato plausibile, dato che entrambi i gruppi idrossi sono attivati (affiancati da gruppi carbossilici o alchinilici). Comunque, in riconoscimento della difficoltà dell'effettuaree sostituzioni su centri stericamente affollati, è stato immaginato un approccio alternativo che presenta un'apertura d'anello diastereoselettiva dell'1,3-diossolanone 8. Questo piano richiede l'addizione promossa da acido di Lewis di bis(trimetilsilil)acetilene ad un acetale attivato17. Perciò, gli stereocentri in C(2) e C(8) sono stati installati attraverso una reazione controllata dallo stereocentro del residuo di acido malico. Entrambi gli approcci sono interessanti, dato che sia 6 che 8 potevano essere facilmente derivati dall'acido L-(S)-malico naturale.
Inoltre, i componenti essenziali in questo approccio dovrebbero essere la reazione di RCM/reazione di cross-coupling intramolecolare Pd-catalizzata, assistita da silicio per la costruzione del nucleo ossoninico, così come l'installazione diastereoselettiva di un centro stereogenico propargilico nell'intermedio chiave 5.
Gli autori descrivono qui la prima sintesi totale della (+)-brasilenina applicando queste nuove trasformazioni quali elementi chiave strategici18.
La soluzione seemplice a questo problema dovrebbe ricorrere ad una reazione di sostituzione nucleofila. Poiché entrambi gli enantiomeri di 615 e 716 sono disponibili, l'unione dell'etere doppiamente ramificato può procedere in entrambe le direzioni. Questo approccio era considerato plausibile, dato che entrambi i gruppi idrossi sono attivati (affiancati da gruppi carbossilici o alchinilici). Comunque, in riconoscimento della difficoltà dell'effettuaree sostituzioni su centri stericamente affollati, è stato immaginato un approccio alternativo che presenta un'apertura d'anello diastereoselettiva dell'1,3-diossolanone 8. Questo piano richiede l'addizione promossa da acido di Lewis di bis(trimetilsilil)acetilene ad un acetale attivato17. Perciò, gli stereocentri in C(2) e C(8) sono stati installati attraverso una reazione controllata dallo stereocentro del residuo di acido malico. Entrambi gli approcci sono interessanti, dato che sia 6 che 8 potevano essere facilmente derivati dall'acido L-(S)-malico naturale.
Inoltre, i componenti essenziali in questo approccio dovrebbero essere la reazione di RCM/reazione di cross-coupling intramolecolare Pd-catalizzata, assistita da silicio per la costruzione del nucleo ossoninico, così come l'installazione diastereoselettiva di un centro stereogenico propargilico nell'intermedio chiave 5.
Gli autori descrivono qui la prima sintesi totale della (+)-brasilenina applicando queste nuove trasformazioni quali elementi chiave strategici18.
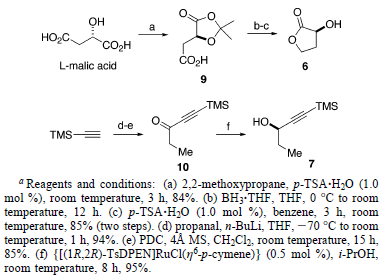 Inoltre, è stato ottenuto 7 molto enantiomericamente arricchito (98.8% ee) con una resa del 95% tramite un'efficiente reazione di idrogenazione (senza idrogeno) del corrispondente chetone 10 in i-PrOH con una quantità catalitica di {[(1R,2R)-TsDPEN]RuCl(η6-p-cimene)}, come sviluppato da Noyori et al19. La purezza enantiomerica di 7 è stata determinata tramite analisi CSP SFC del 3,5-dinitrobenzoato estere derivato.
Con entrambi i precursori enantiomericamente puri in mano, l'obiettivo successivo è stato l'unione di 6 e 7 nell'intermedio 5. Uno studio iniziale sull'attivazione del gruppo idrossile propargilico con Tf2O, seguita dalla sostituzione con 6, è stato senza successo (schema sottostante).
Inoltre, è stato ottenuto 7 molto enantiomericamente arricchito (98.8% ee) con una resa del 95% tramite un'efficiente reazione di idrogenazione (senza idrogeno) del corrispondente chetone 10 in i-PrOH con una quantità catalitica di {[(1R,2R)-TsDPEN]RuCl(η6-p-cimene)}, come sviluppato da Noyori et al19. La purezza enantiomerica di 7 è stata determinata tramite analisi CSP SFC del 3,5-dinitrobenzoato estere derivato.
Con entrambi i precursori enantiomericamente puri in mano, l'obiettivo successivo è stato l'unione di 6 e 7 nell'intermedio 5. Uno studio iniziale sull'attivazione del gruppo idrossile propargilico con Tf2O, seguita dalla sostituzione con 6, è stato senza successo (schema sottostante).
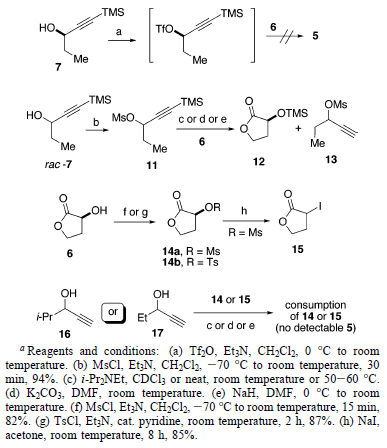 Per identificare il problema con questa trasformazione, è stato preparato il mesilato 11 stabile (per trattamento del rac-7 con
MsCl e Et3N) e quindi sottoposto a formazione di etere con 6 in diverse condizioni. Sfortunatamente, nessuna reazione è avvenuta per niente usando i-Pr2NEt come base. Entrambi i reagenti, 6 e 7, sono stati stabili in queste, ed perfino in quelle più dure, condizioni (50-60 °C e/o puri). Impiegando K2CO3 o NaH come base, sono stati osservati il composto sililato 12 ed il prodotto desililato 13 tramite analisi 1H-NMR. Lo scambio di ruoli tramite attivazione del gruppo idrossile di 6 è stata un'altra opzione. I substrati attivati 14 e 15 sono stati facilmente preparati da 6. Il trattamento dei composti 14 o 15 con gli alcoli propargilici 16 o 17 usando i-Pr2NEt non ha portato ad alcuna reazione. Usando K2CO3 o NaH come base, è stata osservato il completo consumo di 14 o 15, senza una quantità rilevabile di 5.
Apertura d'anello dell'1,3-diossolanone. Il fallimento nel congiungere direttamente le due subunità 6 and 7 ha spostato l'attenzione sulla formazione dell'etere 5 doppiamente ramificato tramite apertura d'anello diastereoselettiva di un 1,3-diossolanone. La sintesi asimmetrica tramite apertura di anello di modelli di acetale chirale promossa da acidi di Lewis fornisce una via di sintesi per la generazione selettiva di alcoli secondari chirali e/o eteri. Durante gli ultimi venti anni, sono stati impiegati con successo molti differenti reagenti organometallici nucleofilici, così come gli acidi di Lewis20. La diastereoselettività di questo processo dipende fortemente dalla struttura dell'acetale, dal solvente, dall'acido di Lewis, così come dal reagente nucleofilo. Tra essi, i composti organometallici acetilenici sono stati impiegati come reagenti nucleofilici per creare un centro stereogenico in posizioni propargiliche con elevata diastereoselettività17. Infatti, è stata profondamente investigata l'apertura d'anello di 1,3-diossolanoni con diversi reagenti nucleofilici ed acidi di Lewis21. Comunque, l'apertura d'anello di un 1,3-diossolanone con reagenti organometallici acetilenici è senza precedenti.
L'1,3-diossolanone 8 è stato scelto per testare questo approccio perché (1) 8 può essere velocemente ottenuto dall'acido naturale L-(S)-malico e (2) la propanale può agire sia da gruppo protettivo sia come parte del composto d'interesse 5 (schema sottostante).
Per identificare il problema con questa trasformazione, è stato preparato il mesilato 11 stabile (per trattamento del rac-7 con
MsCl e Et3N) e quindi sottoposto a formazione di etere con 6 in diverse condizioni. Sfortunatamente, nessuna reazione è avvenuta per niente usando i-Pr2NEt come base. Entrambi i reagenti, 6 e 7, sono stati stabili in queste, ed perfino in quelle più dure, condizioni (50-60 °C e/o puri). Impiegando K2CO3 o NaH come base, sono stati osservati il composto sililato 12 ed il prodotto desililato 13 tramite analisi 1H-NMR. Lo scambio di ruoli tramite attivazione del gruppo idrossile di 6 è stata un'altra opzione. I substrati attivati 14 e 15 sono stati facilmente preparati da 6. Il trattamento dei composti 14 o 15 con gli alcoli propargilici 16 o 17 usando i-Pr2NEt non ha portato ad alcuna reazione. Usando K2CO3 o NaH come base, è stata osservato il completo consumo di 14 o 15, senza una quantità rilevabile di 5.
Apertura d'anello dell'1,3-diossolanone. Il fallimento nel congiungere direttamente le due subunità 6 and 7 ha spostato l'attenzione sulla formazione dell'etere 5 doppiamente ramificato tramite apertura d'anello diastereoselettiva di un 1,3-diossolanone. La sintesi asimmetrica tramite apertura di anello di modelli di acetale chirale promossa da acidi di Lewis fornisce una via di sintesi per la generazione selettiva di alcoli secondari chirali e/o eteri. Durante gli ultimi venti anni, sono stati impiegati con successo molti differenti reagenti organometallici nucleofilici, così come gli acidi di Lewis20. La diastereoselettività di questo processo dipende fortemente dalla struttura dell'acetale, dal solvente, dall'acido di Lewis, così come dal reagente nucleofilo. Tra essi, i composti organometallici acetilenici sono stati impiegati come reagenti nucleofilici per creare un centro stereogenico in posizioni propargiliche con elevata diastereoselettività17. Infatti, è stata profondamente investigata l'apertura d'anello di 1,3-diossolanoni con diversi reagenti nucleofilici ed acidi di Lewis21. Comunque, l'apertura d'anello di un 1,3-diossolanone con reagenti organometallici acetilenici è senza precedenti.
L'1,3-diossolanone 8 è stato scelto per testare questo approccio perché (1) 8 può essere velocemente ottenuto dall'acido naturale L-(S)-malico e (2) la propanale può agire sia da gruppo protettivo sia come parte del composto d'interesse 5 (schema sottostante).
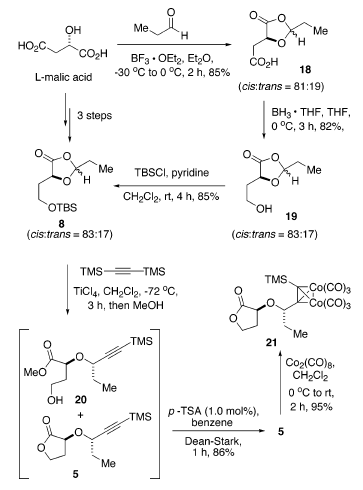 La sintesi di 8 iniziava tramite condensazione dell'acido L-(S)-malico con propanale promossa da BF3·Et2O a dare l'1,3-diossolanone 18 (85%) come una miscela epimerica che favoriva di poco l'isomero cis (cis/trans = 81:19)22. La riduzione selettiva dell'acido carbossilico usando BH3·THF a 0 °C ha portato a 19 (82% yield) seguita da protezione dell'alcole primario appena formato con t-butilclorodimetilsilano, e piridina ha dato 8 con una resa dell'85% (resa totale di ca. 60-65% in tre passaggi da acido L-(S)-malico). La povera selettività nella preparazione del diossolanone 18 è stata inaspettata. Sulla base del precedente in letteratura, la costruzione dell'1,3-diossolanone normalmente favorisce il più termodinamicamente stabile isomero cis con elevata diastereoselettività22. Fortunatamente, questo è stato successivamente dimostrato essere irrilevante. La trasformazione diretta della miscela cis/trans di 18 nell'alcole 19 e nel TBS etere 8 ha mantenuto il rapporto (cis/trans = 83:17). L'intermedio idrossi 1,3-diossolanone 19 è stato labile sia ad acidi sia a basi, quali Et3N, i-Pr2NEt, imidazolo, e N,N-(dimetilamino)piridina, producento l'(S)-α-idrossi-γ-butirrolattone 6. Comunque, portando a termine la riduzione di 18 a 0 °C e la protezione di 19 usando piridina come base, queste trasformazioni potevano essere condotte in modo riproducibile su larga scala.
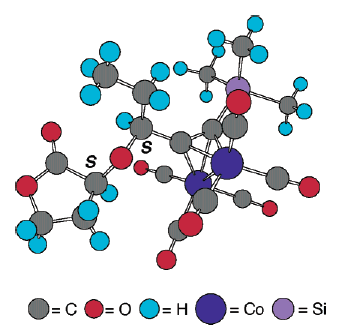
L'apertura d'anello promossa da acido di Lewis di 8 con bis(trimetilsilil)acetilene utilizzava TiCl4 quale acido di Lewis in una procedura simile all'apertura d'anello di modelli acetalici sviluppata da Johnson et al.17a. In modo soddisfacente, l'apertura d'anello del diossolanone 8 è continuata facilmente a dare solo due prodotti dopo quenching con MeOH: il lattone 5 desiderato ed il metil estere 20. Inoltre, il trattamento della miscela grezza con una quantità catalitica di acido p-toluensolfonico in benzene a riflusso per 1 h ha dato esclusivamente 5 con una resa dell'86% quale singolo diastereisomero tramite analisi 1H-NMR. La configurazione S della posizone propargilica è stata ulteriormente stabilita tramite conversione di 5 nel dicobalto complesso 21 con Co2(CO)8 con una resa del 95%. La lenta sublimazione di 21 a 65-68 °C (0.1 mmHg) ha dato dei cristalli rosso scuro, la cui completa stereostruttura è stata confermata tramite una diffrazione ai raggi X da cristallo singolo (figura sottostante)23.
La sintesi di 8 iniziava tramite condensazione dell'acido L-(S)-malico con propanale promossa da BF3·Et2O a dare l'1,3-diossolanone 18 (85%) come una miscela epimerica che favoriva di poco l'isomero cis (cis/trans = 81:19)22. La riduzione selettiva dell'acido carbossilico usando BH3·THF a 0 °C ha portato a 19 (82% yield) seguita da protezione dell'alcole primario appena formato con t-butilclorodimetilsilano, e piridina ha dato 8 con una resa dell'85% (resa totale di ca. 60-65% in tre passaggi da acido L-(S)-malico). La povera selettività nella preparazione del diossolanone 18 è stata inaspettata. Sulla base del precedente in letteratura, la costruzione dell'1,3-diossolanone normalmente favorisce il più termodinamicamente stabile isomero cis con elevata diastereoselettività22. Fortunatamente, questo è stato successivamente dimostrato essere irrilevante. La trasformazione diretta della miscela cis/trans di 18 nell'alcole 19 e nel TBS etere 8 ha mantenuto il rapporto (cis/trans = 83:17). L'intermedio idrossi 1,3-diossolanone 19 è stato labile sia ad acidi sia a basi, quali Et3N, i-Pr2NEt, imidazolo, e N,N-(dimetilamino)piridina, producento l'(S)-α-idrossi-γ-butirrolattone 6. Comunque, portando a termine la riduzione di 18 a 0 °C e la protezione di 19 usando piridina come base, queste trasformazioni potevano essere condotte in modo riproducibile su larga scala.
L'apertura d'anello promossa da acido di Lewis di 8 con bis(trimetilsilil)acetilene utilizzava TiCl4 quale acido di Lewis in una procedura simile all'apertura d'anello di modelli acetalici sviluppata da Johnson et al.17a. In modo soddisfacente, l'apertura d'anello del diossolanone 8 è continuata facilmente a dare solo due prodotti dopo quenching con MeOH: il lattone 5 desiderato ed il metil estere 20. Inoltre, il trattamento della miscela grezza con una quantità catalitica di acido p-toluensolfonico in benzene a riflusso per 1 h ha dato esclusivamente 5 con una resa dell'86% quale singolo diastereisomero tramite analisi 1H-NMR. La configurazione S della posizone propargilica è stata ulteriormente stabilita tramite conversione di 5 nel dicobalto complesso 21 con Co2(CO)8 con una resa del 95%. La lenta sublimazione di 21 a 65-68 °C (0.1 mmHg) ha dato dei cristalli rosso scuro, la cui completa stereostruttura è stata confermata tramite una diffrazione ai raggi X da cristallo singolo (figura sottostante)23.
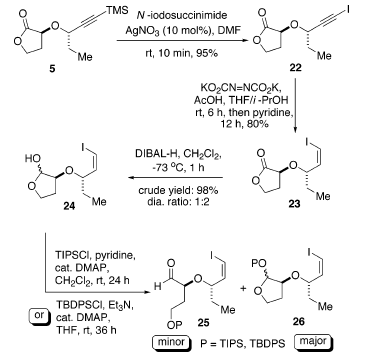 La quantità in tracce del prodotto di sovra-ossidazione può essere rimossa ponendo sotto agitazione la miscela in piridina per 12 h a temperatura ambiente. L'installazione del terzo centro stereogenico in C(8) richiede una reazione di allilazione diastereoselettiva di un'aldeide recante un gruppo idrossile protetto, come 25. Quindi, la riduzione di 23 con DIBAL-H ha portato all'ottenimento del lattolo 24 con una buona resa e rapporto diastereoisomerico di 1:2. Sfortunatamente, tutti i tentativi di proteggere selettivamente l'intermedia idrossi aldeide ottenuta dall'apertura d'anello con TIPSCl o TBDPSCl sono state senza successo. Solo una moderata conversione ed una piccola quantità del prodotto desiderato 25 sono stati ottenuti dopo una difficile separazione dal composto 2626.
In alternativa, il lattone 5 potrebbe essere convertito nell'ammide di Weinreb 28, che ha fornito opzioni alternative per la generazione dell'alcole omoallilico 30. Per esempio, attraverso l'influenza di un gruppo α-alcossi, l'alcole omoallilico 30 potrebbe essere ottenuto sia tramite una riduzione del chetone omoallilico 29 controllata da chelazione o tramite una allilazione di 4 controllata non da chelazione (schema sottostante).
La quantità in tracce del prodotto di sovra-ossidazione può essere rimossa ponendo sotto agitazione la miscela in piridina per 12 h a temperatura ambiente. L'installazione del terzo centro stereogenico in C(8) richiede una reazione di allilazione diastereoselettiva di un'aldeide recante un gruppo idrossile protetto, come 25. Quindi, la riduzione di 23 con DIBAL-H ha portato all'ottenimento del lattolo 24 con una buona resa e rapporto diastereoisomerico di 1:2. Sfortunatamente, tutti i tentativi di proteggere selettivamente l'intermedia idrossi aldeide ottenuta dall'apertura d'anello con TIPSCl o TBDPSCl sono state senza successo. Solo una moderata conversione ed una piccola quantità del prodotto desiderato 25 sono stati ottenuti dopo una difficile separazione dal composto 2626.
In alternativa, il lattone 5 potrebbe essere convertito nell'ammide di Weinreb 28, che ha fornito opzioni alternative per la generazione dell'alcole omoallilico 30. Per esempio, attraverso l'influenza di un gruppo α-alcossi, l'alcole omoallilico 30 potrebbe essere ottenuto sia tramite una riduzione del chetone omoallilico 29 controllata da chelazione o tramite una allilazione di 4 controllata non da chelazione (schema sottostante).
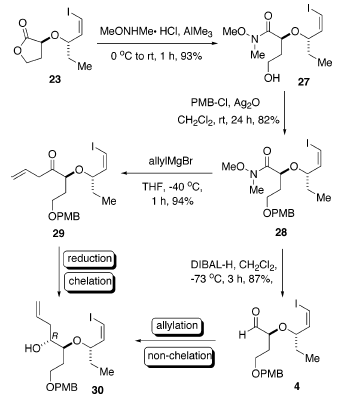 Inoltre, il trattamento di 23 con il sale N,O-dimetilidroamina
idrocloruro in presenza di trimetilalmuminio ha portato all'ammide 27 con una resa del 93%27. Inoltre, la protezione del gruppo idrossile con p-metossibenzile cloruro (PMBCl) nelle condizioni che tollerano la funzionalità vinile ioduro (Ag2O) hanno portato a 28 con una resal dell'82%28. L'ammide 28 potrebbe essere convertita nell'aldeide 4 tramite riduzione con DIBAL-H (resa dell'87%)29 o nel chetone omoallilico 29 attraverso trattamento con allilmagnesio bromuro (resa del 94%)30.
Con entrambi i precursori 4 e 29 in mano, il successivo stadio critico è stato introdurre il terzo centro stereogenico di 1. Gli studi iniziali sulla riduzione del chetone omoallilico di 29 nell'alcole omoallilico 30 sono stilati nella tabella sottostante. L'uso di DIBAL-H quale agente riducente (per tollerarela funzionalità vinile ioduro) ha dato un'eccellente resa chimica (voce 1), ma con diastereoselettività non soddisfacente, favorendo leggermente l'indesiderato isomero S. Tutti i tentativi di aumentare la selettività attraverso l'impiego di MgBr2 e ZnI2 sono stati vani (voci 2 e 3)31. La LS-Selectride ha portato alla formazione dell'indesiderato isomero S (voce 4).
Inoltre, il trattamento di 23 con il sale N,O-dimetilidroamina
idrocloruro in presenza di trimetilalmuminio ha portato all'ammide 27 con una resa del 93%27. Inoltre, la protezione del gruppo idrossile con p-metossibenzile cloruro (PMBCl) nelle condizioni che tollerano la funzionalità vinile ioduro (Ag2O) hanno portato a 28 con una resal dell'82%28. L'ammide 28 potrebbe essere convertita nell'aldeide 4 tramite riduzione con DIBAL-H (resa dell'87%)29 o nel chetone omoallilico 29 attraverso trattamento con allilmagnesio bromuro (resa del 94%)30.
Con entrambi i precursori 4 e 29 in mano, il successivo stadio critico è stato introdurre il terzo centro stereogenico di 1. Gli studi iniziali sulla riduzione del chetone omoallilico di 29 nell'alcole omoallilico 30 sono stilati nella tabella sottostante. L'uso di DIBAL-H quale agente riducente (per tollerarela funzionalità vinile ioduro) ha dato un'eccellente resa chimica (voce 1), ma con diastereoselettività non soddisfacente, favorendo leggermente l'indesiderato isomero S. Tutti i tentativi di aumentare la selettività attraverso l'impiego di MgBr2 e ZnI2 sono stati vani (voci 2 e 3)31. La LS-Selectride ha portato alla formazione dell'indesiderato isomero S (voce 4).
 In seguito, è stata saggiata l'influenza del gruppo α-alcossi sull'allilazione selettiva di 4 tramite un'addizione controllata non da chelazione (tabella più sotto). Usando alliltrimetilsilano quale agente allilante e BF3·Et2O quale acido di Lewis, è stata solo ottenuto un rapporto 26:74 (S/R) dell'alcole 30 con una resa del prodotto isolato del 78%, favorendo il desiderato isomero R (voce 1). Aumentando la quantità di acido di Lewis (4.0 equiv) non si ha avuto un effetto significativo sulla resa o sulla selettività (voce 2)32. È stato anche impiegato l'alliltributilstagno quale reagente allilante33, ma è stata ottenuta una povera selettività anche con questo reagente (voce 3). L'addizione diretta con allilmagnesio bromuro in presenza di ZnCl2 ha portato a 30 con una resa del 91% quale miscela ca. 1:1 di diastereoisomeri (voce 4)34.
In seguito, è stata saggiata l'influenza del gruppo α-alcossi sull'allilazione selettiva di 4 tramite un'addizione controllata non da chelazione (tabella più sotto). Usando alliltrimetilsilano quale agente allilante e BF3·Et2O quale acido di Lewis, è stata solo ottenuto un rapporto 26:74 (S/R) dell'alcole 30 con una resa del prodotto isolato del 78%, favorendo il desiderato isomero R (voce 1). Aumentando la quantità di acido di Lewis (4.0 equiv) non si ha avuto un effetto significativo sulla resa o sulla selettività (voce 2)32. È stato anche impiegato l'alliltributilstagno quale reagente allilante33, ma è stata ottenuta una povera selettività anche con questo reagente (voce 3). L'addizione diretta con allilmagnesio bromuro in presenza di ZnCl2 ha portato a 30 con una resa del 91% quale miscela ca. 1:1 di diastereoisomeri (voce 4)34.
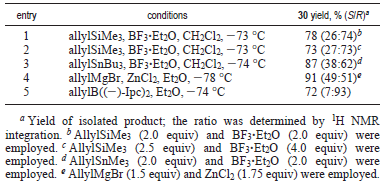 La generazione con successo di 30 è stata ottenuta infine tramite impiego del reagente allilborano chirale, ((-)-Ipc)2Ballile,
sviluppato da Brown et al.35. Il trattamento di 4 con il reagente generato in situ da (+)-B-clorodiisopinocamfeilborano
[(+)-DIP-Cl] ed allilmagnesio bromuro ha portato a 30 con una resa del 72% con una diastereoselettività di 93:7 (voce 5). Questo reagente è stato mostrato che esibisce un forte bias di selettività intrinseca. Quindi, l'allilazione con ((-)-Ipc)2Ballile, derivato dal (-)-α-pinene, dovrebbe dare la corrispondente configurazione R in posizione omoallilica35. Un aumento significativo nella resa (89%) e nella selettività (>97:3) nell'allilazione è stato ottenuto in condizioni senza sale di Mg2+ a -100 °C (schema sottostante)35b.
La generazione con successo di 30 è stata ottenuta infine tramite impiego del reagente allilborano chirale, ((-)-Ipc)2Ballile,
sviluppato da Brown et al.35. Il trattamento di 4 con il reagente generato in situ da (+)-B-clorodiisopinocamfeilborano
[(+)-DIP-Cl] ed allilmagnesio bromuro ha portato a 30 con una resa del 72% con una diastereoselettività di 93:7 (voce 5). Questo reagente è stato mostrato che esibisce un forte bias di selettività intrinseca. Quindi, l'allilazione con ((-)-Ipc)2Ballile, derivato dal (-)-α-pinene, dovrebbe dare la corrispondente configurazione R in posizione omoallilica35. Un aumento significativo nella resa (89%) e nella selettività (>97:3) nell'allilazione è stato ottenuto in condizioni senza sale di Mg2+ a -100 °C (schema sottostante)35b.
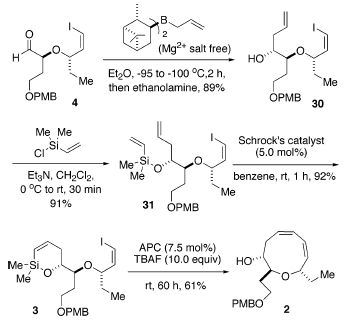 La successiva sililazione dell'alcole secondario con clorodimetilvinilsilano ha portato al vinil silil etere 31 con una resa del 91%. Qusto sililvinil eter è stato sottoposto ad una reazione di RCM usando il complesso di molibdeno di Schrock [(CF3)2MeCO]2Mo(=CHCMe2Ph)(=NC6H3-2,6-i-Pr2) come catalizzatore36. Usando un carico del 5.0 mol % di catalizzatore a temperatura ambiente in benzene, la chiusura d'anello è andata a completezza efficientemente in 1 h porta a 3 con una resa eccellente (92%).
La decisiva reazione di cross-coupling intramolecolare di 3 è stata portata a termine nelle condizioni ottimizzate stabilite in precedenza: 7.5 mol % di [allilPdCl]2 quale catalizzatore e 10 equiv di una soluzione 1.0 M di TBAF quale attivatore usando un'addizione tramite siringa-pompa14. La reazione è continuata senza difficoltà a dare il corrispondente etere a nove membri 2 con una resa del 61%. Sorprendentemente, non è stata osservata alcuna influenza significitiva o della catena laterale ingombrata o del gruppo etile.
La successiva sililazione dell'alcole secondario con clorodimetilvinilsilano ha portato al vinil silil etere 31 con una resa del 91%. Qusto sililvinil eter è stato sottoposto ad una reazione di RCM usando il complesso di molibdeno di Schrock [(CF3)2MeCO]2Mo(=CHCMe2Ph)(=NC6H3-2,6-i-Pr2) come catalizzatore36. Usando un carico del 5.0 mol % di catalizzatore a temperatura ambiente in benzene, la chiusura d'anello è andata a completezza efficientemente in 1 h porta a 3 con una resa eccellente (92%).
La decisiva reazione di cross-coupling intramolecolare di 3 è stata portata a termine nelle condizioni ottimizzate stabilite in precedenza: 7.5 mol % di [allilPdCl]2 quale catalizzatore e 10 equiv di una soluzione 1.0 M di TBAF quale attivatore usando un'addizione tramite siringa-pompa14. La reazione è continuata senza difficoltà a dare il corrispondente etere a nove membri 2 con una resa del 61%. Sorprendentemente, non è stata osservata alcuna influenza significitiva o della catena laterale ingombrata o del gruppo etile.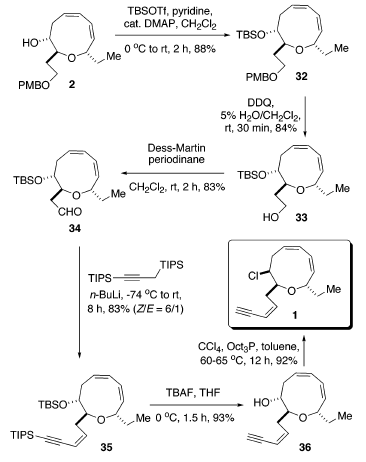 Per evitare la potenziale eliminazione di HCl, l'installazione del cloruro è stata scelta come passaggio finale. L'elaborazione della catena laterale eninica è iniziata tramite protezione del gruppo idrossile con TBSOTf usando piridina ed una quantità catalitica di DMAP portando a 32 con una resa dell'88%. La deprotezione del gruppo PMB con DDQ in CH2Cl2/H2O (19/1)37 ha portato a 33 efficacemente con una resa dell'84%. L'ossidazione del 33 con il periodinano di Dess-Martin38 ha portato alla formazione dell'aldeide 34 con una resa dell'83% (resa totale del 61% dal prodotto di coupling 2). L'olefinazione tipo Peterson come descritta da Corey et al.39 è stata impiegata per introdurre la richiesta catena laterale Z eninica. Quindi, il trattamento di 34 con l'1,3-bis(triisopropil)propino litiato a bassa temperatura, seguito da lento riscaldamento della miscela a temperatura ambiente, ha prodotto l'enino 35 con una resa dell'83% con una selettività geometrica di ca. 6:1 Z/E. I tentativi di aumentare la selettività geometrica attraverso impiego del MgBr2 ha portato al fallimento. Soprattutto, l'eliminazione del gruppo β-alcossi a formare l'aldeide α,β-insatura è stata osservata tramite analisi 1H-NMR [segnali caratteristici: 9.55 (d, J = 8.0, 1H, CHO); 6.78 (dd, J = 15.5, 4.5, 1 H, Hβ); 6.27 (dd, J = 16.0, 8.0, 1 H, Hα)]40. Di conseguenza, la deprotezione del gruppo TBS, così come del gruppo TIPS, con una soluzione 1.0 M di TBAF in THF ha fornito l'idrossi enino 36 con una resa del 93%.
Infine, l'inversione del gruppo idrossile 8R nel cloruro 8S usando CCl4/(n-Oct)3P in toluene11m a 60-65 °C ha completato la prima sintesi totale della (+)-brasilenina (1). i dati fisici e spettroscopici per il campione sintetico sono abbastanza identici in tutti gli aspetti [mp 37-38 °C (lit. 36-37 °C), 1H-NMR, 13C-NMR, IR, e [α]24D = +228.0 (c = 1.08, CHCl3)]; lit. +216 (c
= 0.017, CHCl3) a quelli riportati per la (+)-brasilenina naturale7a.
Discussione
Sebbene la sintesi della (+)-brasilenina fosse stata intrapresa per testare l'applicabilità della tandem RCM/cross-coupling diretto da silicio su un obiettivo sintetico impegnativo, questa trasformazione è avvenuta, felicemente, senza difficoltà. Sulla basi di precedenti studi nei laboratori degli autori, le condizioni ottimizzate per ognuna delle due reazione potevano essere direttamente applicate al substrato più complesso 31 con eccellenti risultati. Inoltre, la manovra chiave strategica provocherà solo un minore commento, e, invece, cederà il riflettore ai giocatori di supporto assegnati per creare i centri stereogenici nella (+)-brasilenina.
Formazione dell'intermedio 5. Il fallimento nel creare un etere doppiamente ramificato tramite le classiche reazioni di sostituzione necessitava lo sviluppo di una strategia alternativa che potesse controllare l'introduzione del centro propargilico da un materiale enantiopuro preesistente. L'apertura diastereoselettiva dell'1,3-diossolanone derivato dall'acido malico ha offerto una soluzione interessante.
Negli studi iniziali sull'apertura d'anello promossa da acidi di Lewis di 8 con bis(trimetilsilil)acetilene, è stato trovato che il TMSOTf era instabile per questa reazione, dando miscele complesse insieme con la deprotezione del diossolanone. Il titanio tetracloruro è stato il più efficace promotore. Inoltre, la procedura di workup influenzava molto la distribuzione dei prodotti. Per esempio, il workup acquoso diretto produceva diversi componenti, come 37a-c e 5 con 37b quale prodotto maggioritario, mentre 20 e 5 veniva ottenuti dopo quenching con metanolo (schema sottostante).
Per evitare la potenziale eliminazione di HCl, l'installazione del cloruro è stata scelta come passaggio finale. L'elaborazione della catena laterale eninica è iniziata tramite protezione del gruppo idrossile con TBSOTf usando piridina ed una quantità catalitica di DMAP portando a 32 con una resa dell'88%. La deprotezione del gruppo PMB con DDQ in CH2Cl2/H2O (19/1)37 ha portato a 33 efficacemente con una resa dell'84%. L'ossidazione del 33 con il periodinano di Dess-Martin38 ha portato alla formazione dell'aldeide 34 con una resa dell'83% (resa totale del 61% dal prodotto di coupling 2). L'olefinazione tipo Peterson come descritta da Corey et al.39 è stata impiegata per introdurre la richiesta catena laterale Z eninica. Quindi, il trattamento di 34 con l'1,3-bis(triisopropil)propino litiato a bassa temperatura, seguito da lento riscaldamento della miscela a temperatura ambiente, ha prodotto l'enino 35 con una resa dell'83% con una selettività geometrica di ca. 6:1 Z/E. I tentativi di aumentare la selettività geometrica attraverso impiego del MgBr2 ha portato al fallimento. Soprattutto, l'eliminazione del gruppo β-alcossi a formare l'aldeide α,β-insatura è stata osservata tramite analisi 1H-NMR [segnali caratteristici: 9.55 (d, J = 8.0, 1H, CHO); 6.78 (dd, J = 15.5, 4.5, 1 H, Hβ); 6.27 (dd, J = 16.0, 8.0, 1 H, Hα)]40. Di conseguenza, la deprotezione del gruppo TBS, così come del gruppo TIPS, con una soluzione 1.0 M di TBAF in THF ha fornito l'idrossi enino 36 con una resa del 93%.
Infine, l'inversione del gruppo idrossile 8R nel cloruro 8S usando CCl4/(n-Oct)3P in toluene11m a 60-65 °C ha completato la prima sintesi totale della (+)-brasilenina (1). i dati fisici e spettroscopici per il campione sintetico sono abbastanza identici in tutti gli aspetti [mp 37-38 °C (lit. 36-37 °C), 1H-NMR, 13C-NMR, IR, e [α]24D = +228.0 (c = 1.08, CHCl3)]; lit. +216 (c
= 0.017, CHCl3) a quelli riportati per la (+)-brasilenina naturale7a.
Discussione
Sebbene la sintesi della (+)-brasilenina fosse stata intrapresa per testare l'applicabilità della tandem RCM/cross-coupling diretto da silicio su un obiettivo sintetico impegnativo, questa trasformazione è avvenuta, felicemente, senza difficoltà. Sulla basi di precedenti studi nei laboratori degli autori, le condizioni ottimizzate per ognuna delle due reazione potevano essere direttamente applicate al substrato più complesso 31 con eccellenti risultati. Inoltre, la manovra chiave strategica provocherà solo un minore commento, e, invece, cederà il riflettore ai giocatori di supporto assegnati per creare i centri stereogenici nella (+)-brasilenina.
Formazione dell'intermedio 5. Il fallimento nel creare un etere doppiamente ramificato tramite le classiche reazioni di sostituzione necessitava lo sviluppo di una strategia alternativa che potesse controllare l'introduzione del centro propargilico da un materiale enantiopuro preesistente. L'apertura diastereoselettiva dell'1,3-diossolanone derivato dall'acido malico ha offerto una soluzione interessante.
Negli studi iniziali sull'apertura d'anello promossa da acidi di Lewis di 8 con bis(trimetilsilil)acetilene, è stato trovato che il TMSOTf era instabile per questa reazione, dando miscele complesse insieme con la deprotezione del diossolanone. Il titanio tetracloruro è stato il più efficace promotore. Inoltre, la procedura di workup influenzava molto la distribuzione dei prodotti. Per esempio, il workup acquoso diretto produceva diversi componenti, come 37a-c e 5 con 37b quale prodotto maggioritario, mentre 20 e 5 veniva ottenuti dopo quenching con metanolo (schema sottostante).
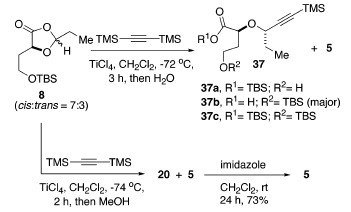 Specialmente, ogni componente includeva solo un singolo diastereoisomero, indicando che il processo di apertura d'aello era altamente diastereoselettivo. La conversione di 20 nel desiderato lattone era facilmente ottenuta tramite trattamento con imidazolo in CH2Cl2 fornendo 5 con una resa del 73% dopo 24 h. Comunque, usando una quantità catalitica di acido p-toluenesolfonico in benzene a riflusso ha dato 5 più efficacemente.
Diastereoselettività dell'apertura di anello di 8. L'apertura d'anello di acetali modello, con reagenti acetilenici debolmente nucleofilici, quali il bis(trimetilsilil)acetilene, presumibilmente reagisce nella regione SN1 del continuum meccanicistico (cioè, tramite apertura d'anello assistita da acido di Lewis prima dell'attacco nucleofilico)20h. In questi casi, l'induzione asimmetrica è stata razionalizzata tramite l'invocazione della regola di Cram come applicato allo ione intermedio ossocarbenio17b. Effettivamente, il fatto che un singolo diastereoisomero 5 sia stato ottenuto da una miscela cis/trans di 8 indicava fermamente che (1) il meccanismo dell'apertura d'anello dell'1,3-diossolanone procedeva anche attraverso uno ione intermedio ossocarbenio e (2) l'elevata diastereoselettività era totalmente controllata dal centro stereogenico del residuo acido L-(S)-malico21a. Vale la pena notare che il più labile legame C-OCO nell'1,3-diossolanone puà facilitare la formazione di un ione intermedio ossocarbenio reattivo, particolarmente in presenza di un forte acido di Lewis quale il TiCl4.
Una giustificazione per la convergente diastereoselettività è formulata nella figura sottostante.
Specialmente, ogni componente includeva solo un singolo diastereoisomero, indicando che il processo di apertura d'aello era altamente diastereoselettivo. La conversione di 20 nel desiderato lattone era facilmente ottenuta tramite trattamento con imidazolo in CH2Cl2 fornendo 5 con una resa del 73% dopo 24 h. Comunque, usando una quantità catalitica di acido p-toluenesolfonico in benzene a riflusso ha dato 5 più efficacemente.
Diastereoselettività dell'apertura di anello di 8. L'apertura d'anello di acetali modello, con reagenti acetilenici debolmente nucleofilici, quali il bis(trimetilsilil)acetilene, presumibilmente reagisce nella regione SN1 del continuum meccanicistico (cioè, tramite apertura d'anello assistita da acido di Lewis prima dell'attacco nucleofilico)20h. In questi casi, l'induzione asimmetrica è stata razionalizzata tramite l'invocazione della regola di Cram come applicato allo ione intermedio ossocarbenio17b. Effettivamente, il fatto che un singolo diastereoisomero 5 sia stato ottenuto da una miscela cis/trans di 8 indicava fermamente che (1) il meccanismo dell'apertura d'anello dell'1,3-diossolanone procedeva anche attraverso uno ione intermedio ossocarbenio e (2) l'elevata diastereoselettività era totalmente controllata dal centro stereogenico del residuo acido L-(S)-malico21a. Vale la pena notare che il più labile legame C-OCO nell'1,3-diossolanone puà facilitare la formazione di un ione intermedio ossocarbenio reattivo, particolarmente in presenza di un forte acido di Lewis quale il TiCl4.
Una giustificazione per la convergente diastereoselettività è formulata nella figura sottostante.
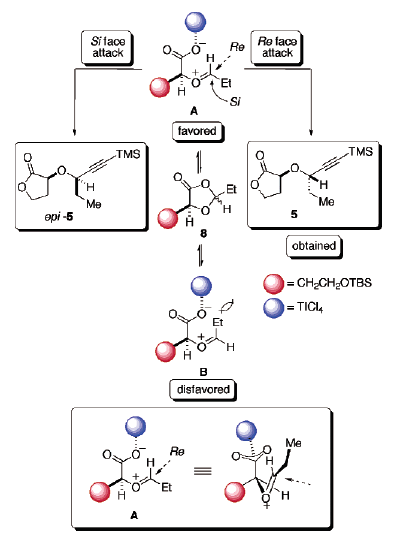 L'equilibrazione degli intermedi A e B potrebbe presumibilmente essere ottenuta facilmente in presenza di un forte acido di Lewis acid21a. Per minimizzare la repulsione dipolo-dipolo, lo ione ossocarbenio ed il carbossilato coordinato al Ti sono disposti in una conformazione antiperiplanare. Inoltre, lo ione A ossocarbenio E derivato dall'apertura di anello è proposto essere quello termodinamicamente favorito. Questo è a causa della ridotta interazione sterica tra il residuo acido malico ed il gruppo etile. Inoltre, l'attacco nucleofilo dalla faccia Re evita una significativa repulsione sterica dal gruppo R ingombrato (CH2CH2OTBS), portando quindi al desiderato diastereoisomero 5.
Allilborazione dell'aldeide 4. L'impossibilità di realizzare una diastereoselettività controllata da substrato nell'addizione nucleofila di idruri al chetone 29 o di reagenti organometallici all'aldeide 4 suggerì l'uso del reagente di Brown ((-)-Ipc)2Ballile, che esibisce un bias intrinseco sconvolgente35. La più altra resa (89%) e diastereoselettività (>97:3) sono stati raggiunti nelle condizioni senza sali di magnesio a -100 °C (schema sottostante)35b.
L'equilibrazione degli intermedi A e B potrebbe presumibilmente essere ottenuta facilmente in presenza di un forte acido di Lewis acid21a. Per minimizzare la repulsione dipolo-dipolo, lo ione ossocarbenio ed il carbossilato coordinato al Ti sono disposti in una conformazione antiperiplanare. Inoltre, lo ione A ossocarbenio E derivato dall'apertura di anello è proposto essere quello termodinamicamente favorito. Questo è a causa della ridotta interazione sterica tra il residuo acido malico ed il gruppo etile. Inoltre, l'attacco nucleofilo dalla faccia Re evita una significativa repulsione sterica dal gruppo R ingombrato (CH2CH2OTBS), portando quindi al desiderato diastereoisomero 5.
Allilborazione dell'aldeide 4. L'impossibilità di realizzare una diastereoselettività controllata da substrato nell'addizione nucleofila di idruri al chetone 29 o di reagenti organometallici all'aldeide 4 suggerì l'uso del reagente di Brown ((-)-Ipc)2Ballile, che esibisce un bias intrinseco sconvolgente35. La più altra resa (89%) e diastereoselettività (>97:3) sono stati raggiunti nelle condizioni senza sali di magnesio a -100 °C (schema sottostante)35b.
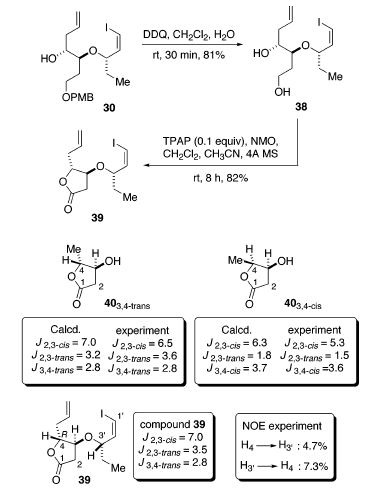 Il trattamento di 30 con DDQ in CH2Cl2/H2O (19/1) ha dato il diolo 38 con una resa dell'81%.
La successiva ossidazione è stata condotta con una quantità catalitica di tetrapropilammonio perrutenato (TPAP) e N-metilmorfolina-N-ossido fornendo il lattone 39 con una resa dell'82%42. Negli spettri 1H-NMR di 39, le costanti di accoppiamento J2,3-cis, J2,3-trans, e J3,4-trans sono di 7.0, 3.5, e 2.8 Hz, rispettivamente. Questi dati sono ben in accordo con quelli calcolati (7.0, 3.2, e 2.8 Hz) e a quelli determinati sperimentalmente (6.5, 3.6, 2.8 Hz) per il 403,4-trans. Soprattutto, sono stati anche osservati forti aumenti NOE tra HC(4) e HC(3'
Il trattamento di 30 con DDQ in CH2Cl2/H2O (19/1) ha dato il diolo 38 con una resa dell'81%.
La successiva ossidazione è stata condotta con una quantità catalitica di tetrapropilammonio perrutenato (TPAP) e N-metilmorfolina-N-ossido fornendo il lattone 39 con una resa dell'82%42. Negli spettri 1H-NMR di 39, le costanti di accoppiamento J2,3-cis, J2,3-trans, e J3,4-trans sono di 7.0, 3.5, e 2.8 Hz, rispettivamente. Questi dati sono ben in accordo con quelli calcolati (7.0, 3.2, e 2.8 Hz) e a quelli determinati sperimentalmente (6.5, 3.6, 2.8 Hz) per il 403,4-trans. Soprattutto, sono stati anche osservati forti aumenti NOE tra HC(4) e HC(3' . Un aumento del 4.7% di HC(3'
. Un aumento del 4.7% di HC(3'