Mercaptano
2020-06-11 22:44
Buonasera, o buongiorno, a seconda dei vostri ritmi circadiani, oggi si parla di Neuraminidasi Influenzali, ai meno conosciute come eso-alfa-sialidasi influenzali (EC 3.2.1.19).
Si tratta di una discussione aperta a consigli e che spero di aggiornare al più presto con un bel po' di informazioni. Intanto lascio qui lo spunto che Claudio G. mi ha dato quando si stava discutendo di tutt'altro.
Lascio tutti i messaggi intonsi:
Io:
(l'ho disegnato perché i programmi di disegno molecolare fanno le frecce di spostamento elettronico bruttissime)
Ma no, ma no... un po' di smanettamento e le frecce su Chemscketch escono bellissime, lo sto utilizzando proprio ora per il progetto di maturità.
Scusate il parziale OT.
--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
Claudio:
[quote='Mercaptano Ha scritto:']
Ma no, ma no... un po' di smanettamento e le frecce su Chemscketch escono bellissime, lo sto utilizzando proprio ora per il progetto di maturità.
Scusate il parziale OT.
Uh anvedi, buono. Io son solito usare MarvinSketch, che davvero fa delle frecce bruttissime. Però proverò, grazie per il consiglio 
Uh, è una sialidasi quella? Scusa se ti faccio un appunto, ma in quell'intermedio non dovrebbe entrare direttamente acqua, con attacco sul C1 e rottura del legame alla tirosina? Cioè se ho capito bene tu intendi far attaccare nel passaggio successivo il carbocatione (che formi con gli spostamenti indicati) dall'ossidrile, quindi un attacco Sn1. Però nota anche (probabilmente lo avrai già considerato, ma nel dubbio...) che è pure possibile un meccanismo Sn2 (vedi il caso scuola della catalisi da lisozima), e che di norma nelle glicoside idrolasi è preferito, perché evita la formazione di cariche nette 
--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
Io:
Uh anvedi, buono. Io son solito usare MarvinSketch, che davvero fa delle frecce bruttissime. Però proverò, grazie per il consiglio
Uh, è una sialidasi quella? Scusa se ti faccio un appunto, ma in quell'intermedio non dovrebbe entrare direttamente acqua, con attacco sul C1 e rottura del legame alla tirosina? Cioè se ho capito bene tu intendi far attaccare nel passaggio successivo il carbocatione (che formi con gli spostamenti indicati) dall'ossidrile, quindi un attacco Sn1. Però nota anche (probabilmente lo avrai già considerato, ma nel dubbio...) che è pure possibile un meccanismo Sn2 (vedi il caso scuola della catalisi da lisozima), e che di norma nelle glicoside idrolasi è preferito, perché evita la formazione di cariche nette
Sì, hai perfettamente ragione, secondo l'ultima proposta accreditata sarebbe così, però ci sono un po' di cose che non tornano dai paper che ho letto.
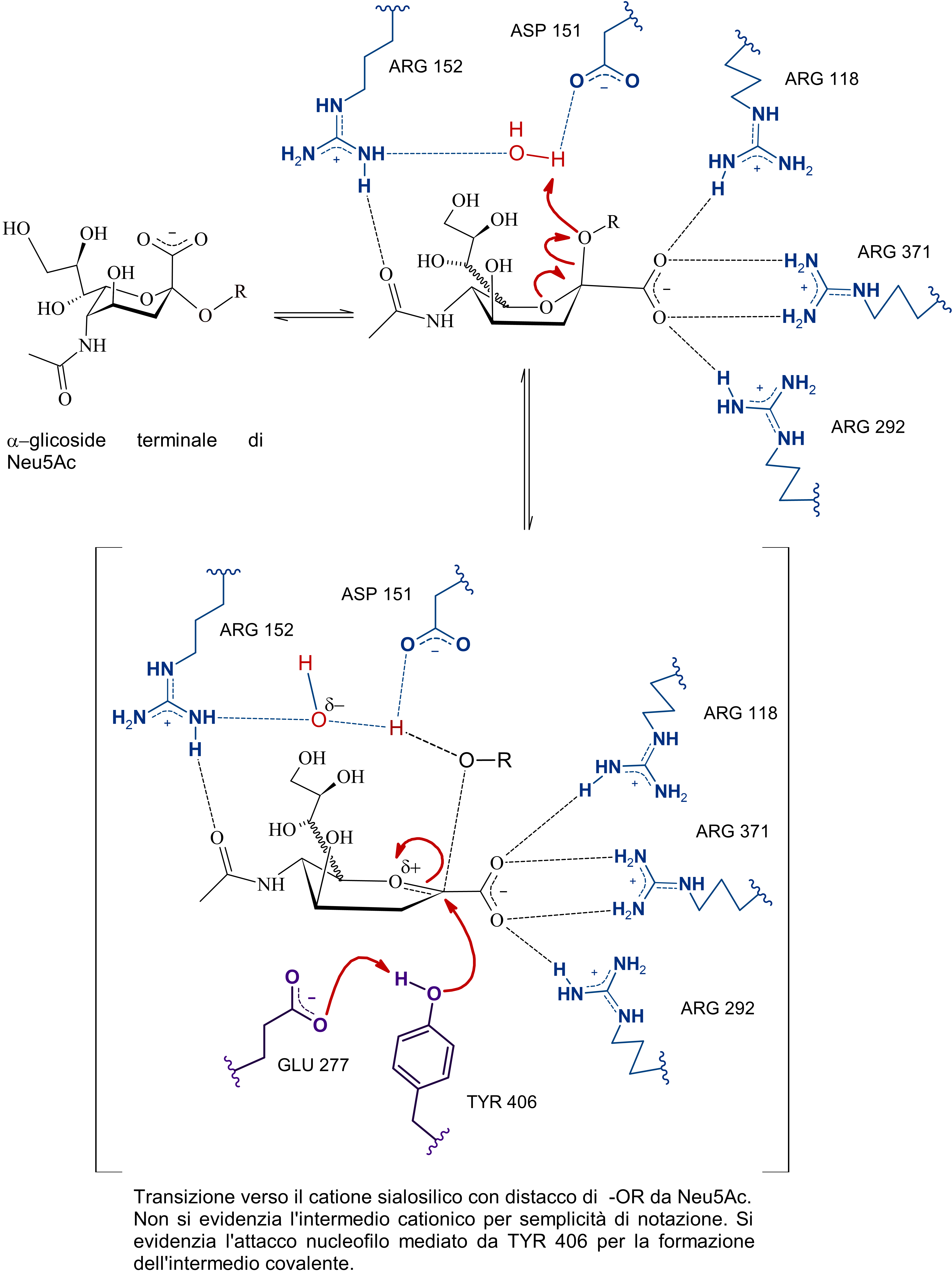
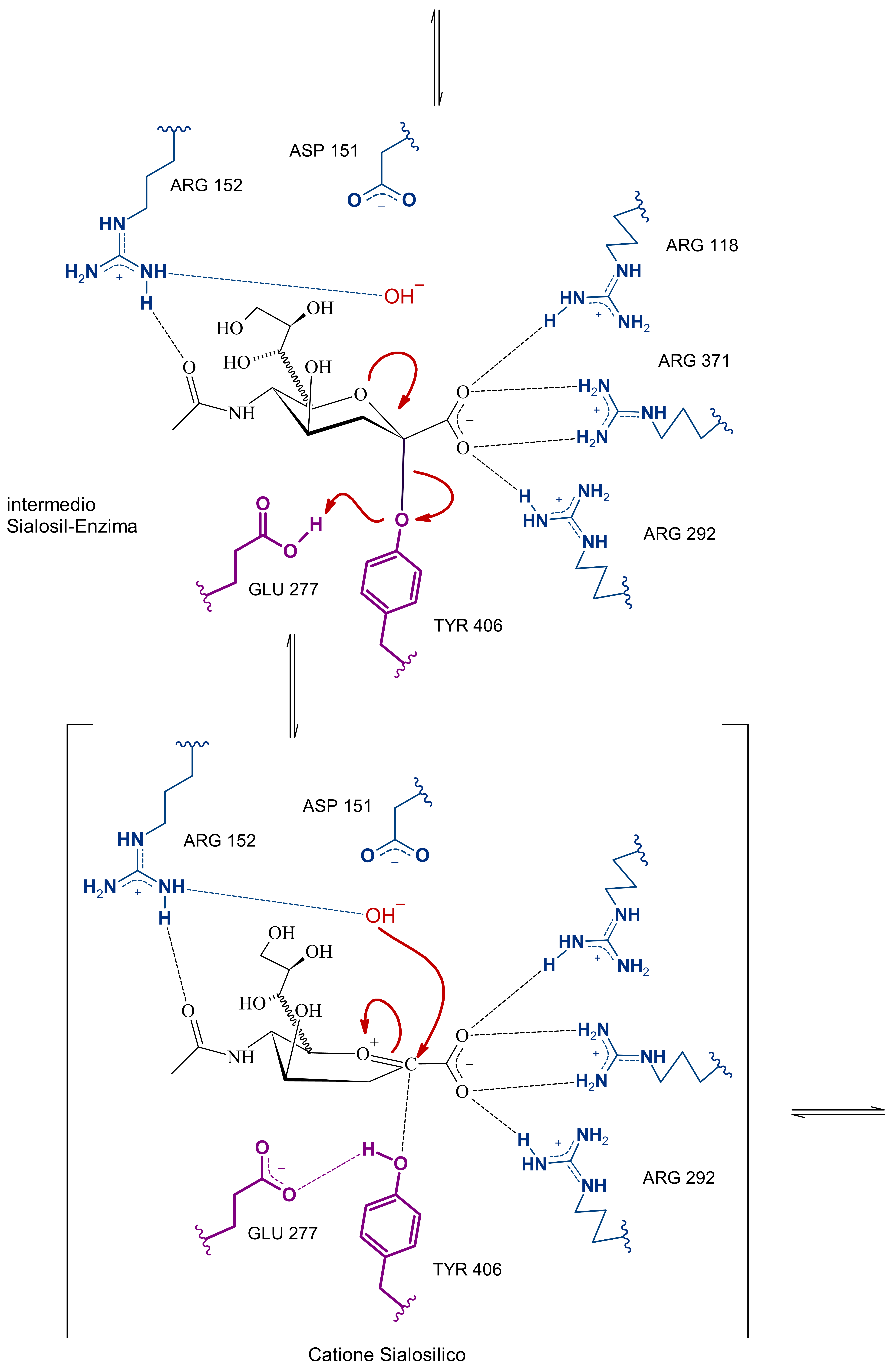
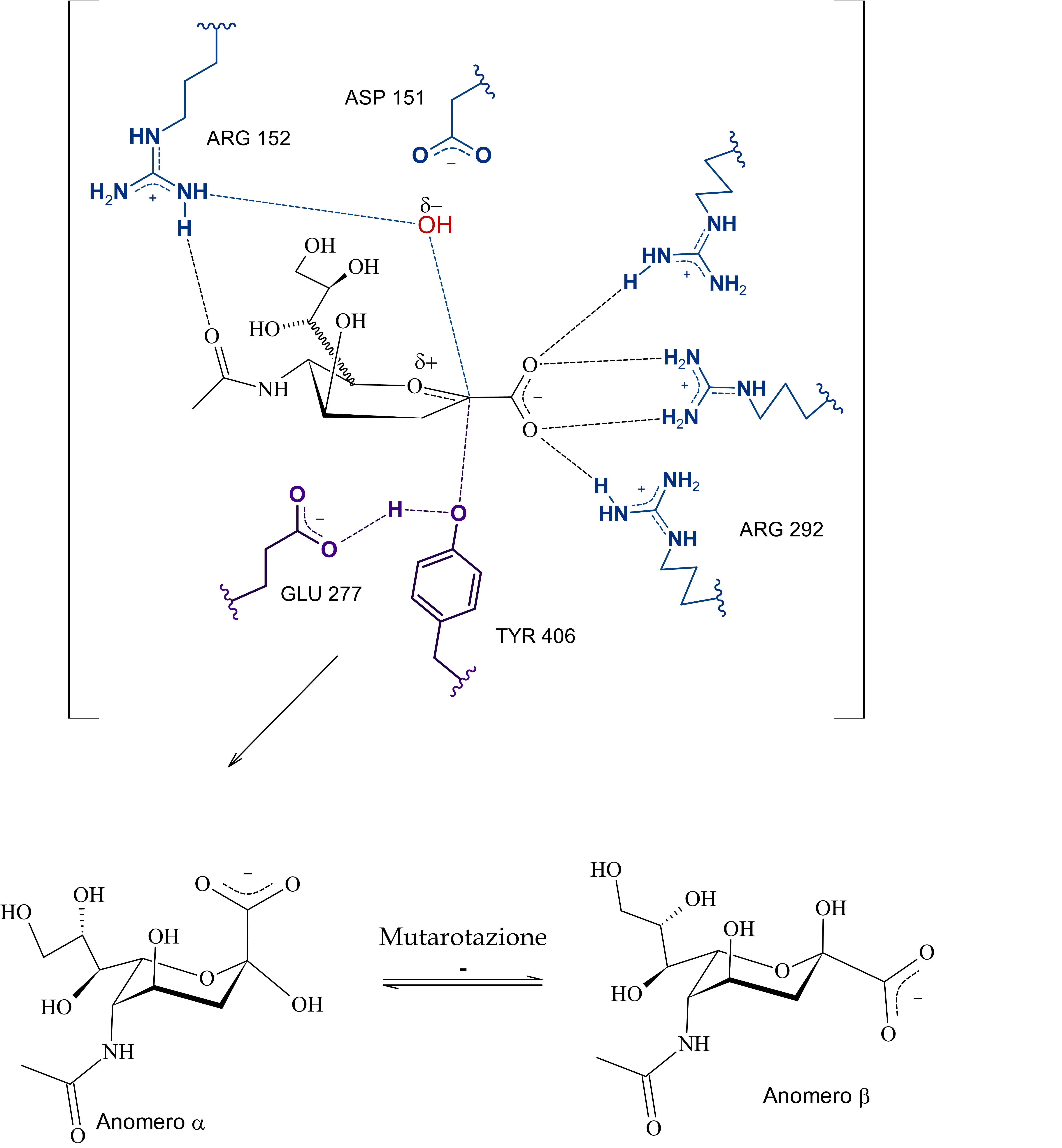
Si tratta di una sialidasi virale, in particolare, che a quanto pare dalle cristallografie a raggi X e dalle simulazioni ha di mezzo anche un osso-carbocatione sialosilico stabilizzato dal sito attivo e che per la sua formazione prevede un aspartato protonato (ASP 151) che funga da catalizzatore acido-base.
Il problema è che non sono presenti evidenze (o forse sono io che davvero non riesco a comprenderlo ahah) di come possa venir protonato questo ASP 151, dunque ho scritto sia una ipotesi basata sul primo paper di Taylor et al., quella che vedi sopra, dove a mediare l'inizio della catalisi sono ARG152 e ASP151 che orientano l'acqua in maniera tale da permettere l'attacco da parte di un doppietto dell'ossigeno del legame glicosidico nei confronti del protone.
Poi dunque dopo la formazione del catione sialosilico, l'allontanamento della catena oligosaccaridica -OR, e la formazione dell'intermedio covalente, l'ossidrile rimasto, stabilizzato dall'arginina e da altri residui, attacca il carbonio anomerico permettendo la rottura del legame tra tirosina e acido sialico (dopo quindi c'è il distacco del prodotto e la sua mutarotazione in anomero beta), in quest'ultima serie di passaggi, ho incorporato anche le evidenze di un intermedio covalente, non citate da Taylor in quanto non era stata ancora dimostrata questa teoria.
Subito sotto la proposta personale (con citazioni a supporto e presa molto con le pinze, perché non sono certo in grado di validarla sperimentalmente), ho riportato l'ipotesi di Mark Von Itzstein et al. secondo cui, come dici tu è una molecola d'acqua a mediare la rottura del complesso sialosil-enzima e nella fasi precedenti, invece, è l'ASP151 protonato a fornire il protone per l'inizio della catalisi.
Per il discorso sul lisozima, iniziando a studiare le sialidasi influenzali sono rimasto interdetto anch'io, poi ho letto meglio e praticamente non si riformerebbe il catione sialosilico (che invece figura in tutte le proposte anche dopo la rottura dell'intermedio covalente).
P.S. se magari apro un'altra discussione, si può fare una bella chiacchierata sulle proposte di meccanismo per le sialidasi, che sono davvero affascinanti. Ho anche pronte una valanga di immagini 
EDIT: ora che ci penso, si avrebbe anche una inversione di configurazione con l'approccio Sn2, o sbaglio? Mentre qui il prodotto dovrebbe rimanere nell'anomero alfa
Se è davvero troppo OT, magari qualcuno può splittare la discussione??
--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
E qui si conclude lo scambio di messaggi, chiunque si voglia unire alla discussione è benvenuto, domattina mi ci metto e carico parte del commento che ho scritto per la maturità, intanto lascio le immagini del meccanismo di reazione secondo Mark Von Itzstein, dell' Institute for Glycomics della Griffith University, in Australia. Riporto il meccanismo in ordine nelle tre immagini da sinistra verso destra.
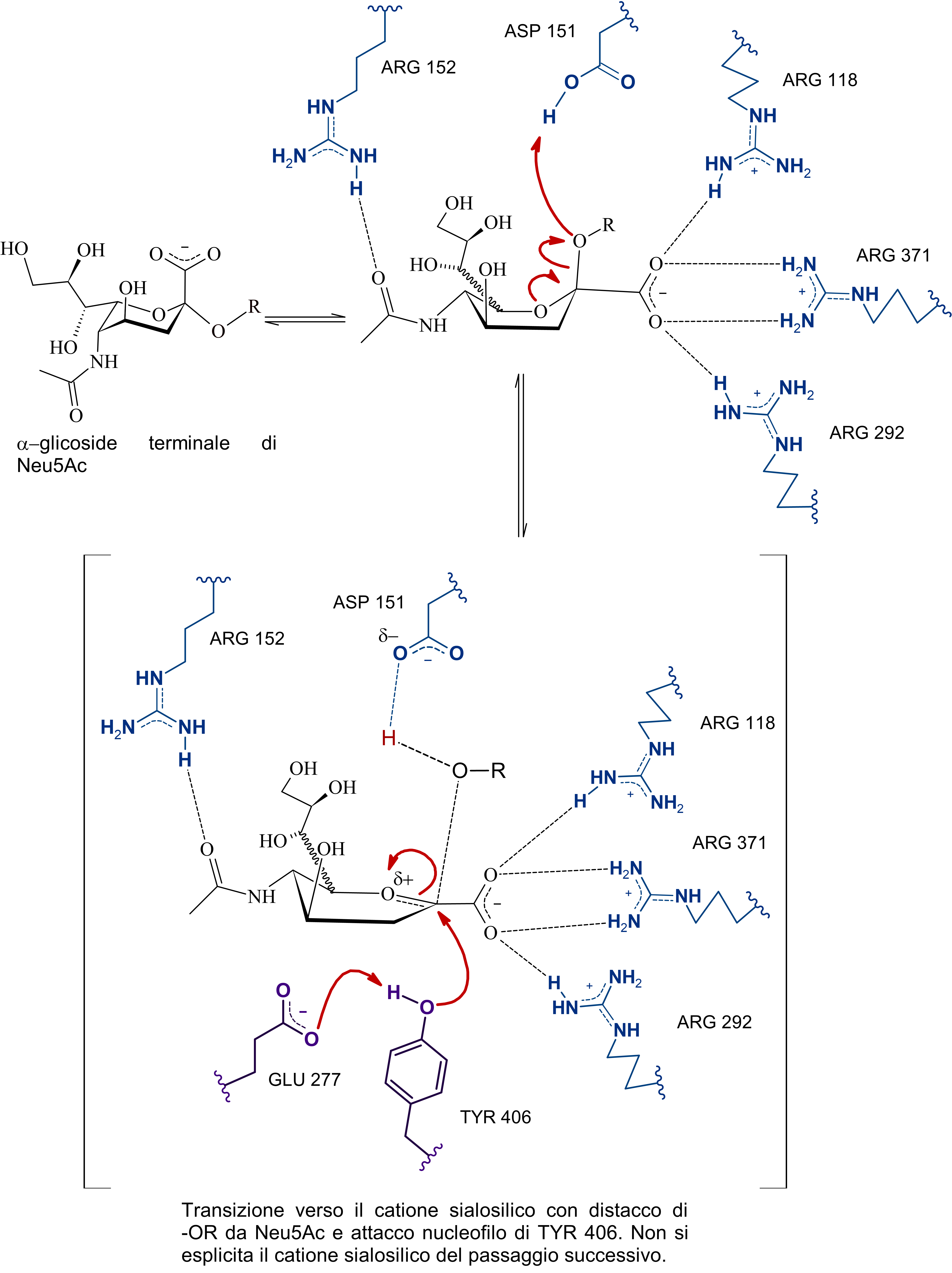
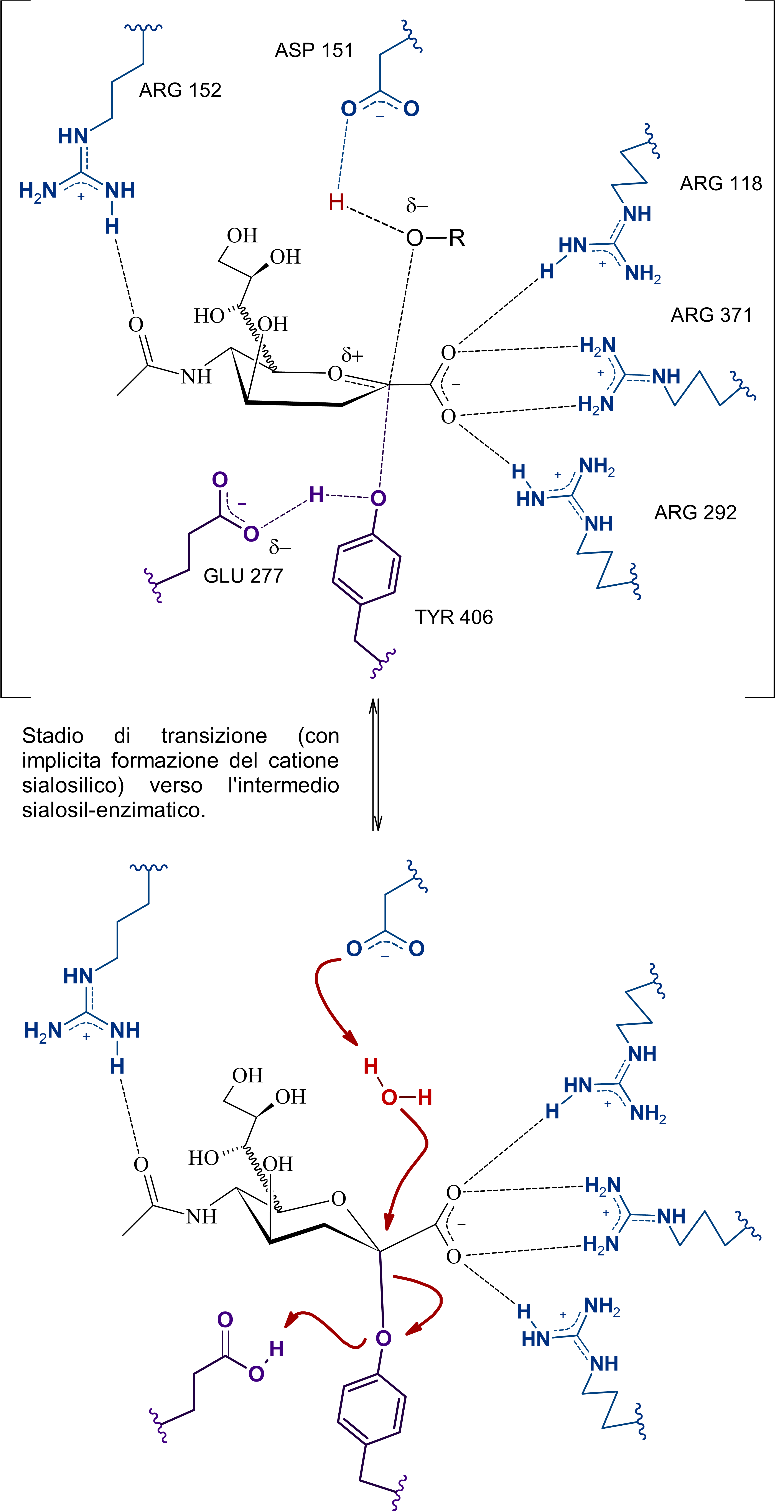
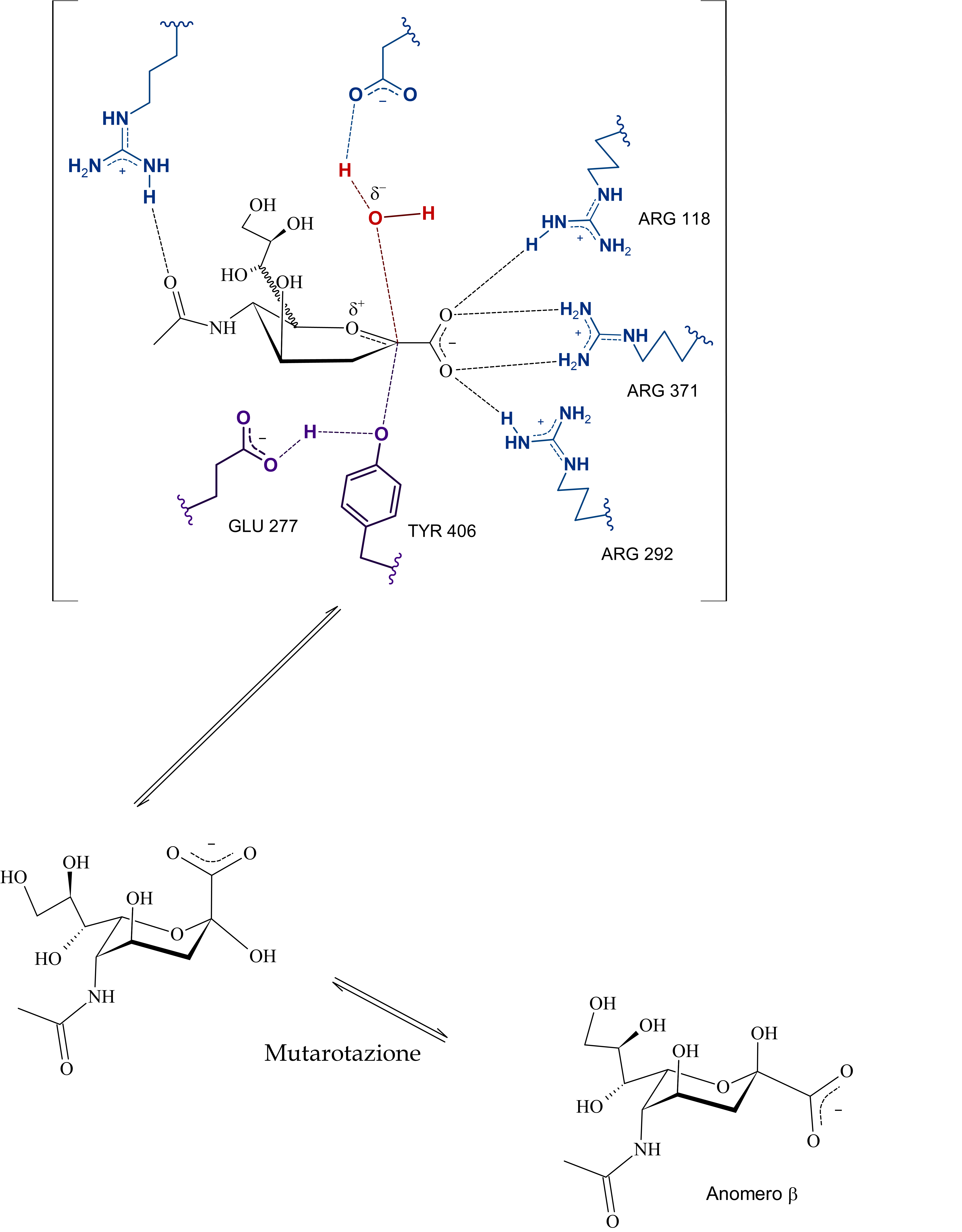
Il modello qui riportato si avvale per la descrizione dello stereocentro del C6 di legame stereochimico indefinito, in quanto la rappresentazione dell’anomero α sarebbe complicata e confusionaria con i metodi grafici utilizzati. Nota: nell'ultimo passaggio ho omesso la formazione di un complesso con conformazione a barca dell'anomero alfa di acido sialico con l'enzima (che poi è il complesso enzima-prodotto finale da cui si distacca il composto in conformazione a sedia)
I seguenti utenti ringraziano Mercaptano per questo messaggio: ClaudioG., luigi_67, TrevizeGolanCz, myttex