Hegelrast
2012-08-28 21:23
Dopo aver chiesto il benestare ai vertici del forum  , vorrei iniziare qui a presentarvi alcune sintesi che ho svolto nell’ultimo annetto circa, usando come reagente di partenza il fenilacetonitrile, o benzil cianuro che dir si voglia (PhCH2CN). Questa sostanza, che purtroppo è controllata come precursore di stupefacenti (ma la cui detenzione non è illegale al di sotto di certi quantitativi, mi risulta) è un liquido paglierino non molto viscoso, con un odore estremamente caratteristico e difficile da descrivere, che non ha nulla a che vedere con il benzonitrile né con l’acido fenilacetico. Il fenilacetonitrile è molto interessante principalmente perchè permette una vastissima gamma di reazioni per la sua “triplice” reattività: per il gruppo nitrile che è di per sé molto versatile, per la presenza dell’anello aromatico e per l’attivazione della posizione benzilica grazie al gruppo elettron-attrattore.
, vorrei iniziare qui a presentarvi alcune sintesi che ho svolto nell’ultimo annetto circa, usando come reagente di partenza il fenilacetonitrile, o benzil cianuro che dir si voglia (PhCH2CN). Questa sostanza, che purtroppo è controllata come precursore di stupefacenti (ma la cui detenzione non è illegale al di sotto di certi quantitativi, mi risulta) è un liquido paglierino non molto viscoso, con un odore estremamente caratteristico e difficile da descrivere, che non ha nulla a che vedere con il benzonitrile né con l’acido fenilacetico. Il fenilacetonitrile è molto interessante principalmente perchè permette una vastissima gamma di reazioni per la sua “triplice” reattività: per il gruppo nitrile che è di per sé molto versatile, per la presenza dell’anello aromatico e per l’attivazione della posizione benzilica grazie al gruppo elettron-attrattore.
La prima reazione che vi descrivo è in realtà molto semplice, sia concettualmente che operativamente: la nitrazione del fenilacetonitrile con isolamento selettivo del p-nitro derivato. La nitrazione avviene a freddo con miscela solfonitrica (sostituzione elettrofila aromatica) senza particolari difficoltà e l’isolamento del prodotto voluto si effettua tramite ricristallizzazione.

Reagenti:
- fenilacetonitrile
- acido nitrico 65%
- acido solforico 96%
- etanolo 95°
Procedimento:
In un pallone a fondo tondo da 250 mL a due o tre colli munito di termometro, agitatore magnetico e imbuto gocciolatore, si versano 27.5 mL di acido nitrico concentrato e si aggiunge lentamente un uguale volume di acido solforico concentrato.


Si fa partire l’agitazione e si immerge il pallone in un bagno di ghiaccio, lasciando raffreddare la miscela fino a 10°C.
Nell’imbuto gocciolatore si versano 10 g (9.8 mL, 85.4 mmol) di fenilacetonitrile e si inizia il a gocciolarlo lentamente in modo da mantenere la temperatura intorno ai 10°C (io ci ho messo una mezz'oretta). E' importante non superare mai i 20°C. La miscela risulta omogenea di colore giallastro, leggermente tendente all’arancione (probabilmente per NO2 disciolta). Al termine dell’aggiunta si allontana il bagno di ghiaccio e si lascia sotto agitazione la miscela per una ulteriore ora.


Si versa la soluzione in 120 mL di ghiaccio tritato e si lascia sciogliere il ghiaccio. Precipita una massa pastosa color crema, costituita dal prodotto e dal suo isomero orto, insieme a una piccola quantità di impurezze oleose.

Si filtra la massa (io mi sono dovuto arrangiare con un buon vecchio filtro di carta, che ha funzionato egregiamente), la si lava con acqua e la si “spreme” per eliminare il più possibile i residui oleosi.


La massa pastosa ancora umida viene sciolta in 50 mL di etanolo al 95% bollente. Lasciando raffreddare la soluzione, il p-nitrofenilacetonitrile cristallizza quasi puro in aghetti incolori. Per massimizzare il recupero è opportuno immergere in ghiaccio il contenitore. Si filtrano i cristalli e li si lasciano asciugare all’aria.




Il Vogel a questo punto suggerisce di ricristallizzare una seconda volta da etanolo 80% bollente, ma per i miei scopi la purezza era già più che sufficiente così (vedi sotto).
Osservazioni:
- La resa di p-nitrofenilacetonitrile è di 7.7 g (circa 56%). Il Vogel riporta una resa del 54% dopo la seconda ricristallizzazione da etanolo 80%, ma lavorando su scala dieci volte superiore. Probabilmente se avessi fatto anche quel passaggio avrei ottenuto qualcosa come un 45% di resa.
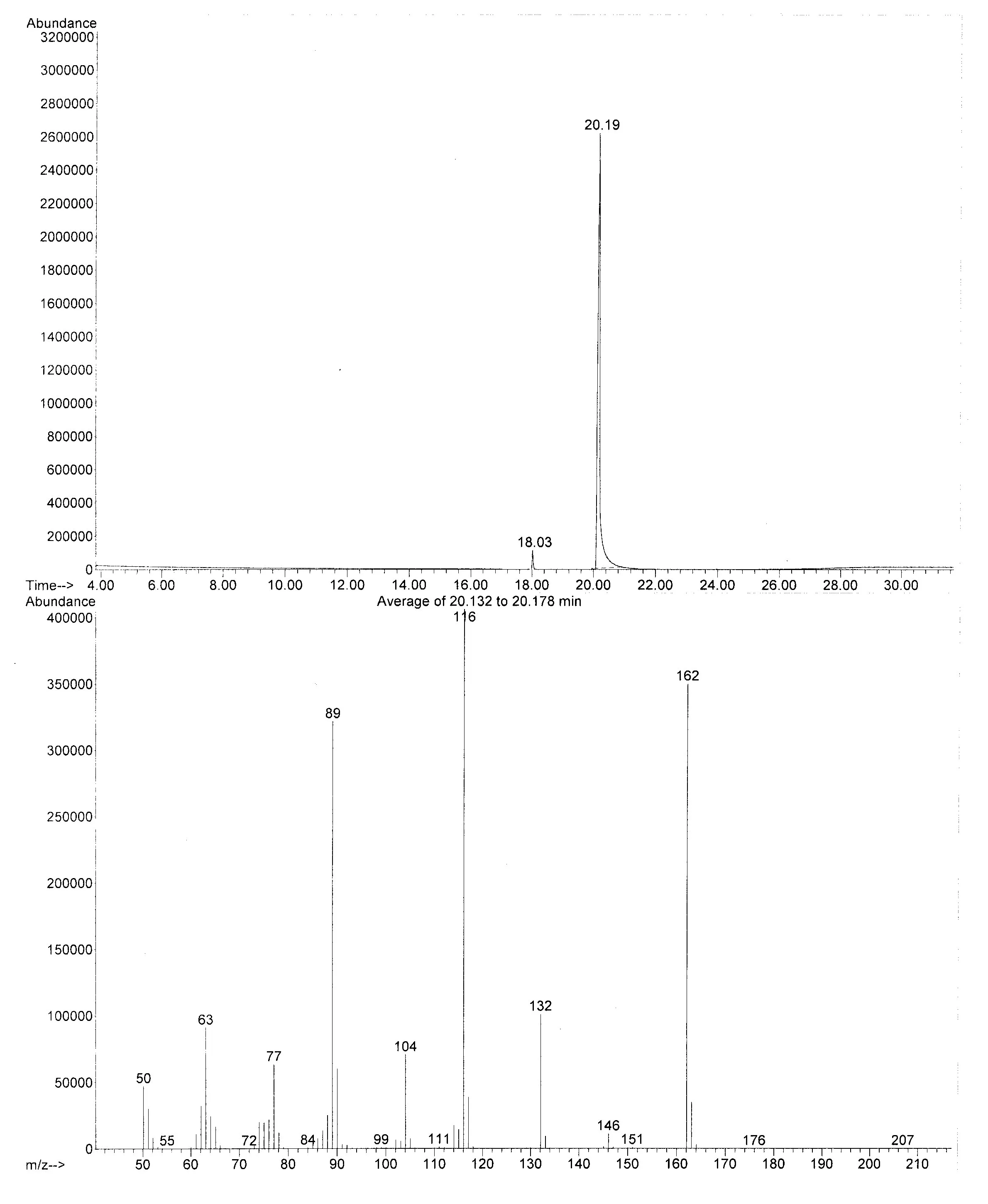
- Il prodotto da me ottenuto ha una purezza superiore al 97% (verificato da GC-MS) e l’unico contaminante risulta essere l’isomero orto.
- Per gli appassionati di analitica strumentale allego una scansione del cromatogramma GC e dello spettro di massa del prodotto ricristallizzato: il picco a 18.03 min è l’isomero orto, il picco a 20.19 min è il prodotto voluto.
- In mancanza di un sistema di aspirazione è indispensabile operare all’aperto dato che il reagente ha un odore molto intenso e che durante la nitrazione si sviluppa una quantità di ossidi di azoto decisamente percettibile (anche se non elevata).
Alla prossima, probabilmente con una bella reazioncina che sfrutta l'acidità dei protoni benzilici.
I seguenti utenti ringraziano Hegelrast per questo messaggio: quimico, ale93, marco the chemistry, Max Fritz, jobba, thenicktm