quimico
2011-09-21 09:45
Dato che ale93 ha fatto una bella olefinazione secondo il buon vecchio Wittig, mi pare giusto completare il quadro introducendo questa reazione. Una reazione sicuramente importante dal punto di vista sintetico ma per certi versi datata e superata (in alcuni casi) da diverse varianti che per lo più usano ancora composti del fosforo ma che usano anche altri composti (silicio o zolfo ad esempio). Ma visto che non siamo qui a metter lo smalto alle mancurie, iniziamo.
 La reazione di Wittig permette la preparazione di un alchene tramite reazione di un aldeide o un chetone con un'ilide generata da un sale di fosfonio.
La geometria dell'alchene risultante dipende dalla reattività dell'ilide.
Se R è Ph, allora l'ilide è stabilizzata e non ha la stessa reattività di quando R = alchile. Le ilidi stabilizzate danno alcheni a stereochimica (E) mentre ilidi non stabilizzate portano ad alcheni a stereochimica (Z) (in questo ultimo caso solitamente si usa la variante di Wittig-Horner*).
Meccanismo della reazione di Wittig
L'addizione di un'ilide al carbonile è postulata condurre prima ad un intermedio zwitterionico o betaina, la quale dovrebbe quindi chiudere a formare un intermedio ciclico a quattro termini, un ossafosfetane.
L'esistenza della betaina non è stata completamente stabilita, sebbene la sua intermediazione giochi un ruolo importante nella modificazione di Schlosser**.
Le betaine possono essere stabilizzate tramite sali di litio portando a sottoprodotti; tuttavia, basi utili per la reazione di Wittig sono ad esempio: NaH, NaOMe, NEt3.
La reazione di Wittig permette la preparazione di un alchene tramite reazione di un aldeide o un chetone con un'ilide generata da un sale di fosfonio.
La geometria dell'alchene risultante dipende dalla reattività dell'ilide.
Se R è Ph, allora l'ilide è stabilizzata e non ha la stessa reattività di quando R = alchile. Le ilidi stabilizzate danno alcheni a stereochimica (E) mentre ilidi non stabilizzate portano ad alcheni a stereochimica (Z) (in questo ultimo caso solitamente si usa la variante di Wittig-Horner*).
Meccanismo della reazione di Wittig
L'addizione di un'ilide al carbonile è postulata condurre prima ad un intermedio zwitterionico o betaina, la quale dovrebbe quindi chiudere a formare un intermedio ciclico a quattro termini, un ossafosfetane.
L'esistenza della betaina non è stata completamente stabilita, sebbene la sua intermediazione giochi un ruolo importante nella modificazione di Schlosser**.
Le betaine possono essere stabilizzate tramite sali di litio portando a sottoprodotti; tuttavia, basi utili per la reazione di Wittig sono ad esempio: NaH, NaOMe, NEt3.
 La driving force è la formazione di un davvero stabile fosfinossido:
La driving force è la formazione di un davvero stabile fosfinossido:
 Le ilidi reattive danno una rapida reazione e un subitanea e veloce apertura d'anello a dare alcheni (Z):
Le ilidi reattive danno una rapida reazione e un subitanea e veloce apertura d'anello a dare alcheni (Z):
 * La variante di Wittig-Horner o reazione di Horner-Wadsworth-Emmons
* La variante di Wittig-Horner o reazione di Horner-Wadsworth-Emmons
 La reazione di aldeiddi o chetoni con ilidi di fosforo stabilizzate (carbanioni fosfonati) conduce ad olefine con un'eccellente selettività E.
Meccanismo della reazione di Wittig-Horner
È simile al meccanismo della Wittig. La stereochimica è decisa dal controllo sterico, dove l'approccio antiperiplanare del carbanione al carbonio del gruppo carbonilico è favorito quando il più piccolo idrogeno aldeidico si eclissa rispetto alla funzionalità fosforanilica ingrombrante. Questo pone il gruppo estereo syn rispetto al gruppo R aldeidico, ma il nascente alchene assume un'orientazione E di questi gruppo dopo rotazione a formare l'ossafosfetano. Poiché il controione litio non interferisce con la formazione dell'ossafosfetano, è possibile usare il BuLi, ma anche NaH e NaOMe sono utili basi per la formazione dell'ilide. Il risultante sottoprodotto fosfato è prontamente separato dai prodotti desiderati semplicemente tramite lavaggio con acqua.
La reazione di aldeiddi o chetoni con ilidi di fosforo stabilizzate (carbanioni fosfonati) conduce ad olefine con un'eccellente selettività E.
Meccanismo della reazione di Wittig-Horner
È simile al meccanismo della Wittig. La stereochimica è decisa dal controllo sterico, dove l'approccio antiperiplanare del carbanione al carbonio del gruppo carbonilico è favorito quando il più piccolo idrogeno aldeidico si eclissa rispetto alla funzionalità fosforanilica ingrombrante. Questo pone il gruppo estereo syn rispetto al gruppo R aldeidico, ma il nascente alchene assume un'orientazione E di questi gruppo dopo rotazione a formare l'ossafosfetano. Poiché il controione litio non interferisce con la formazione dell'ossafosfetano, è possibile usare il BuLi, ma anche NaH e NaOMe sono utili basi per la formazione dell'ilide. Il risultante sottoprodotto fosfato è prontamente separato dai prodotti desiderati semplicemente tramite lavaggio con acqua.


 ** Modificazione di Schlosser
** Modificazione di Schlosser
 La modificazione di Schlosser della Wittig permette la formazione selettiva di alcheni E tramite l'uso di un eccesso di sali di litio durante lo step di addizione dell'ilide e i successivi step di deprotonazione/protonazione.
Meccanismo della modificazione di Schlosser
L'effetto dei sali di litio è che le betaine intermedie non reagiscono ulteriormente. Queste litio betaine che sono abbastanza stabili possono essere deprotonate. La deprotonazione è uguale alla perdita di un centro stereogenico. L'uso di un donatore di protoni stericamente ingombrato porta quindi ad una litio betaina trans. La reazione procede "normalmente", se il litio è scambiato col potassio.
La modificazione di Schlosser della Wittig permette la formazione selettiva di alcheni E tramite l'uso di un eccesso di sali di litio durante lo step di addizione dell'ilide e i successivi step di deprotonazione/protonazione.
Meccanismo della modificazione di Schlosser
L'effetto dei sali di litio è che le betaine intermedie non reagiscono ulteriormente. Queste litio betaine che sono abbastanza stabili possono essere deprotonate. La deprotonazione è uguale alla perdita di un centro stereogenico. L'uso di un donatore di protoni stericamente ingombrato porta quindi ad una litio betaina trans. La reazione procede "normalmente", se il litio è scambiato col potassio.

L'olefinazione (Z)-stereospecifica può anche essere condotta usando la reazione di Still-Gennari (Still, W.C.; Gennari, C., TETRAHEDRON LETTERS, 1983, 24, 4405). Usando fosfonati con gruppi elettron-attrattori (in questo caso trifluoroetile) in combinazione con condizioni fortemente dissocianti (KHMDS e 18-corona-6 in THF) può essere ottenuta una produzione di alchene esclusivamente Z.
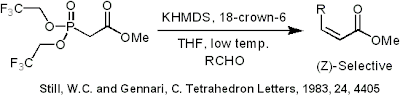 Still-Gennari phosphonate reaction
Still-Gennari phosphonate reaction


I seguenti utenti ringraziano quimico per questo messaggio: Rusty, ale93, Max Fritz
 Ci si legge
Ci si legge  I veri chimici sono quelli che ascoltato e cercano di migliorarsi sempre. Non scordarlo mai!!!
I veri chimici sono quelli che ascoltato e cercano di migliorarsi sempre. Non scordarlo mai!!! 















 Con piccoli controioni (Li) e in solventi apolari si formerà un chelato, che porterà ad uno stato di transizione chiuso.
Con piccoli controioni (Li) e in solventi apolari si formerà un chelato, che porterà ad uno stato di transizione chiuso. Con grossi controioni (K) e in solventi polari, diventa possibile uno stato di transizione aperto.
Con grossi controioni (K) e in solventi polari, diventa possibile uno stato di transizione aperto.






 Ne so abbastanza... Mi hanno indottrinato mica male
Ne so abbastanza... Mi hanno indottrinato mica male 
